Click here for a PDF copy of the User Guide
______________________________________________________________________________________________
Brief Theory of Operation
·
The thermodynamic
properties for all the substances in the ChemReaX database are based on
standard state conditions. Gas species are ideal gases at the standard pressure
of 1 bar. Solute standard states are based on molality, and solutions are at 1
molal concentration at the standard pressure of 1 bar.
·
Since all gas
species are ideal gases at the standard pressure, the corresponding fugacity
coefficients are the low-pressure value of 1. Therefore, the equilibrium constant
on a partial-pressure basis, Kp, is nearly equal to the thermodynamic
equilibrium constant K.
·
All aqueous
solutions are assumed to be dilute, therefore the thermodynamic equilibrium
constant Km written in terms of molality (moles/kg) is assumed to be
nearly equal to the equilibrium constant Kc written in terms of
molarity (moles/L).
·
All aqueous solutions are
assumed to be dilute, therefore the thermodynamic equilibrium constant Km
written in terms of molality (moles/kg) is assumed to be nearly equal to the
equilibrium constant Kc written in terms of molarity (moles/L).
·
All chemical reactions
considered here are assumed to take place at constant temperature and
pressure under typical laboratory conditions.
·
The
standard free-energy change of a reaction at temperature T is: ∆rGo(T) = ∆rHo(T) - T * ∆So(T), where ∆rHo(T) is the net
change in the enthalpies of formation and ∆So(T) is the change in
entropy of the sytem. ∆rGo(T) is an ideal
theoretical property of a reaction at a specified temperature and quantifies
the free-energy change corresponding to the transformation from reactants at
standard states to products at standard states at that temperature.
·
The
reaction free-energy change at temperature T is: ∆rG(T) = ∆rGo(T) + R * T * ln Q,
where R is the gas constant, T is the temperature and Q is the reaction quotient. For a
generic reaction aA + bB <--> cC + dD,
∆rGo(T) = c∆fGco(T) + d∆fGdo(T)
- a∆fGao(T)
- b∆fGbo(T), where the subscript
f implies free-energy of formation.
·
The reaction free-energy change ∆rG(T) = ∆rH(T) - T *
∆S(T)
relates the idealized standard
free-energy change
∆rGo(T)
(which corresponds to
standard conditions for all reactants and products) to the actual
conditions consisting of specific non-standard concentrations or partial
pressures for the reactants and products. While
∆rGo(T) is a
static property of the reaction and a snapshot of the reaction in time,
∆rG(T) is a
dynamic state function of the reaction system that varies over time as the
reaction proceeds in either direction, and is the slope of the free energy G(T)
plotted against the extent of the reaction. Chemical reactions are spontaneous
in the direction of decreasing free energy (by way of increasing the system
entropy and/or decreasing the system enthalpy),
which essentially maximizes the entropy of the universe (system +
surroundings).
o
If
∆rG(T) is negative, the reaction
proceeds spontaneously to the right and increases product concentrations or
partial pressures at the expense of reactants, increasing Q and bringing
∆rG(T) closer to zero; if positive, the
reaction proceeds spontaneously to the left and increases reactant quantities,
decreasing Q and again bringing ∆rG(T)
closer to zero. In both cases, the free energy, G(T), decreases spontaneously. ∆rG(T) is zero if and when the
reaction reaches equilibrium and G(T) is at a minimum -- then neither the
forward nor the reverse reaction has a tendency to occur.
o If the
system entropy decreases in a reaction, then for the change to be spontaneous,
some of the system energy must escape
as heat and increase
the entropy of the surroundings in order to compensate for the reduction in the
system entropy.
∆rG(T) is the portion of the enthalpy change of the
system that is available for doing non-expansion work after increasing the
entropy of the surroundings. If the system entropy increases in a reaction, then
the free energy available for doing non-expansion work is more than the enthalpy
change since some additional energy from the surroundings (upto
T * ∆S(T))
can be drawn into the system.
o
Note that endothermic reactions
(with positive enthalpy change for the system, resulting in an entropy reduction
of the surroundings) can still be spontaneous if the system entropy increases
enough to make
∆rG(T) negative.
·
At equilibrium, ∆rGo(T) = -R * T * ln
Qeq = -R * T *
ln K. The equilibrium constant K is independent of the initial compositions of
the reactants and products, and depends only on thermodynamic quantities that
are constant in the reaction at the given temperature. K represents the
equilibrium reaction quotient Qeq expressed in terms of partial pressures or
concentrations depending on whether the reaction involves gases or occurs in
solution. A relatively large K indicates that the reaction will move
spontaneously to the right. For a generic reaction aA + bB <--> cC + dD, K
can be written as the proper quotient of partial pressures or molar concentrations
(under assumptions of ideal gases and dilute aqueous solutions):
o K = Kp
= Qeq = (pc(C) * pd(D))/(pa(A) * pb(B))
o K = Kc
= Qeq = ([C]c * [D]d)/([A]a * [B]b)
·
The concentrations (more formally, the activities) of pure
solids and liquids (including solvents) are excluded from the reaction quotient
because they are constant at constant temperature and usually very large
compared to the concentrations of gases and solutions. There are exceptions,
such as when a pure solid is used as a solute in a solution and a pure liquid
is a product of a reaction in small quantities.
o The reaction
quotient Q = exp((∆rG(T) - ∆rGo(T)/(R * T)).
The reaction free-energy change includes the free-energy change of
each reactant/product. As an example of a pure solid, consider a system in which
there is ice at -10C. The vapor pressure (p) of ice remains constant at this
temperature, so that
G(T) - Go(T)
= R * T* ln(p/p0) = 0 for the ice;
therefore the ice has no impact on how the reaction moves towards equilibrium
and can be excluded from the reaction quotient Q. Similarly, the concentration
of liquid water is constant at constant temperature and has no impact on the
progress of the reaction and can be excluded from Q.
o
Rewriting
the reaction free-energy change as ∆rG(T) = R * T * ln Q/Qeq = R * T * ln Q/K,
it is easy to see that any reactant or product which is at constant partial
pressure or concentration throughout a reaction (at the given temperature) will
have no impact on the reaction free-energy change. The partial pressure or
concentration of that reactant/product will simply cancel out in the ratio Q/Qeq and
can therefore be omitted from Q and K.
o
Based on
the above, two assumptions are needed to simulate reactions:
(1) Pure solids and liquids that are excluded from the reaction quotient are not
limiting reactants and have no impact on the reaction progress. (2) The reaction
has at least one gas or solution species (including solids, liquids or gases
acting as solutes and not excluded from the reaction quotient), which drive(s)
the reaction towards equilibrium.
·
Per
Le Chatelier's principle, changes in temperature, concentration or pressure will
shift the reaction equilibrium such that the changes can be absorbed.
o
An increase in reactant
concentrations will move the equilibrium to the right such that the increase can
be consumed, and an increase in product concentrations will move the equilibrium
to the left.
o
An increase in
temperature will move the equilibrium to the right for endothermic reactions and
to the left for exothermic reactions in order to absorb the additional heat
being supplied. A decrease in temperature will have the opposite effect. (See
next bullet below for more insight into the thermodynamics behind this
behavior.)
o
If the pressure
within a reaction vessel is
increased or decreased by confining the gas species to a smaller or larger
volume (when a reaction is at equilibrium otherwise), then the equilibrium
composition of the gases will change such that the total number of gas molecules
are decreased or increased. Equilibrium will shift towards the reactants or
products depending on which direction counteracts the pressure change.
∆rGo(T)
and K are independent of the pressure. As an example (from Atkins and De Paula),
consider the reaction: N2 + 3H2 <---> 2NH3. Using partial pressures, the
equilibrium constant K = p2NH3/(pN2 * p3H2)
= x2NH3/(xN2 * x3H2
* p2)
= Kx/p2 , where the x's are
mole fractions and p is the sum of all the reactant/product partial pressures. Kx
is the part of the equilibrium constant that is based on mole fractions. If
pressure doubles in this example, then Kx should increase by
a factor of 4 in order to maintain a constant value of K, thus changing the
equilibrium composition of the gases. The new mole fractions based on the
increased Kx value can be converted back to
equilibrium partial pressures via multiplying each of them by 2p in this case.
Note that injecting an inert gas into the reaction vessel will not shift the
equilibrium since the partial pressures of all reactants/products will be
unchanged (for ideal gases).
·
Rewriting the free-energy change as -∆rG(T)/T = -∆rH(T)/T +
∆S(T) provides
further insight into the thermodynamics of temperature changes.
-∆rH(T) is the enthalpy increase of the
surroundings, -∆rH(T)/T is the change of entropy of the
surroundings, and ∆S(T) is the change of entropy of
the reaction system. A reaction proceeds in the direction that maximizes the
increase in the total entropy of the universe, which is the sum of the entropy
increases of the surroundings and of the system. When a reaction is exothermic,
the entropy of the surroundings increases since -∆rH(T)/T
is positive for the surroundings (heat flows from system to surroundings);
however, as T increases, the entropy of the surroundings does not increase as
much, and the equilibrium falls less to the right. For en endothermic reaction, -∆rH(T)/T
is negative for the surroundings (heat flows from surroundings to system) and
the entropy of the surroundings decreases; however, as T increases, the entropy
of the surroundings does not decrease as much and the equilibrium falls more to
the right. Since ∆S(T)
changes only minimally as T varies (within some range), nearly the entire
response of the reaction equilibrium to temperature changes is dependent on the
entropy of the surroundings.
· Using a
similar insight, when a liquid
turns into vapor at room temperature, the ∆S(T) of
the system is large enough to allow sufficient ∆rH(T)
to be drawn from the environment (thus decreasing the entropy of the
surroundings by ∆rH(T)/T ) and still
increase the total entropy of the universe. For liquids that need a higher
temperature to evaporate, ∆S(T) is not large enough
and ∆rH(T) must be drawn from the
environment at a higher temperature in order for the entropy drop of the
surroundings to be less than the entropy increase of the system.
·
Patterns of entropy changes in solutions:
o When a solid
solute is added to a solvent, the entropy of the solvent and the solute each
increases in the solution since the motional energy of individual
solute/solvent molecules is more dispersed.
o When a gas solute
is dissolved in a solvent, entropy decreases since a solvated gas has lower
entropy than in the pure gas phase.
o When a solute is
ionized in an aqueous solution, the ions tend to orient the water molecules and
cause the entropy to decrease, especially for small or highly charged ions.
·
For the dissociation of acids and bases, the ionization or
dissociation constants Ka and Kb (determined by
experiment) are related to the thermodynamic standard free-energy change by: ∆rGo(T) = -R * T * ln
Ka/b. Given the standard enthalpy change based on
thermodynamic properties, the entropy change for acid/base dissociation can be
calculated as: ∆So(T) = ∆rHo(T)/T + R * ln Ka/b
·
The law of mass
action states that the rate of a reaction at a constant temperature depends only
on the concentrations of the substances (usually one or more reactants) that
influence the rate. The rate also depends on catalysts which do not appear in
the balanced equation. In practice, the reaction rates -- including the overall
order -- can only be determined by experiment under specific conditions. A
generalized integrated reaction rate model used in ChemReaX is: Rate = k *
[R1]X
* [R2]Y * [R3]Z, where R1, R2 and R3 are the reactants. The constants k, X, Y and Z must be experimentally determined for a given reaction at a given temperature. The overall order of a reaction is X+Y+Z. The rate constant k = A e-Ea/RT, which is the Arrhenius equation, where A is the pre-exponential factor, Ea is the activation energy Ea and T is the temperature. Only the reactants are included in this rate model since the product concentrations do not vary independently and are dependent on the reactant concentrations at every point in time. When ChemReaX simulates a reaction, it is essentially
integrating the differential equations numerically.
·
An acid-base titration
can be used to determine the concentration of an acid or base (the titrand) by
exactly neutralizing it with a strong acid or base (the titrant) of known
concentration. Incremental amounts of the titrant are added to a given amount of
the titrand, and pH is measured at each increment after the solution reaches
equilibrium. The result is plotted as the pH of the solution as a function of
the titrant added. The end point or equivalence point of a titration is the
region of steepest rise/fall in pH, where the titrant has exactly neutralized
the titrand. Either the titrand or the titrant will be a limiting reagent in
these reactions. Titration involves an acid-base equilibrium at each increment
of the titrant volume, where two competing simultaneous equilibria must be
accounted for: the ionization of the titrand (which could be a strong/weak acid
or base) and the water autoionization. The final H(+) and OH(-) concentrations
-- and the pH -- at each titrant increment are an outcome of resolving these
simultaneous equilibria.
o
An additional adjustment to
the pH must be made for the hydrolysis of water by the salts produced by the
neutralization of weak acids/bases. The conjugate acid or base produced by the
neutralization can extract an OH- or H+ from water to form the original weak
base/acid and release additional H+ or OH- into the solution. As a result,
aqueous solutions of salts of weak acids are basic, and aqueous solutions of
salts of weak bases are acidic. ChemReaX graphs the pH (as a function of titrant
volume) with and without hydrolysis in order to demonstrate the difference --
hydrolysis basically accelerates the pH change as the titration approaches the
equivalence point.
o In the
case of polyprotic acids
(strong or weak acids with more than one H+ to lose), there are multiple
equivalence points corresponding to multiple ionizations. ChemReaX simulates
only the first two ionizations of such acids, and ignores any additional
ionizations. Hydrolysis is simulated for both ionizations, but omitted for
clarity in any ionization where the equilibrium constant is less than 1e-9. The
second ionization is assumed to start once the first H+ from the acid has been
completely neutralized, which is a reasonable assumption if the equilibrium
constant of the first ionization is much greater than that of the second
ionization (such that as each individual proton in the aqueous solution is
neutralized by the base, the first ionization will preferentially supply the
next proton). Note that a relatively strong second ionization can depress the pH
at the end of the first ionization to well below the neutral value of 7.
How
to Use ChemReaX to Model/Simulate General Reactions
Click on the "General Reactions" tab, and then:
1. With "Select Ionization Reaction" unchecked:
·
Select reactants and products using the dropdown lists. The dropdown lists
are searchable, so you can type any part of a chemical species formula or state
(crystal, liquid, gas, or aq) or charge (+, -, +2, -2, etc.) to find all
matching items.
o
Up to three reactants and three products can be selected.
o
For
aqueous solutions, choose aqueous species if they are available; otherwise,
choose gas, liquid or solid (crystal) species as solutes.
· Balance
the reaction by entering the correct stoichiometric coefficient for each
reactant or product, or click "Balance the Equation" to automatically perform
the balancing.
·
Use the checkbox to
exclude pure solids and liquids from the reaction quotient calculations.
o
This
is provided as an option if certain pure liquids (usually as products of
reactions and in small quantities) do need to be included in the reaction
quotients. Solids need to be included when they are used as solutes in
solutions.
·
Alternately,
click “Reaction Selector” to choose from a pre-defined list of reactions. This
automatically fills in the reactants and products, balances the equation, and
indicates whether certain pure solids or liquids must be excluded from the
reaction quotient. (This list of pre-defined reactions will be extended over
time.)
·
Enter the initial
composition for reactants and products using the following units:
o
Gases: bars
o
Aqueous ions or
aqueous solutions or pure liquids: moles/L
o
Gases, liquids or
solids (crystals) used as solutes in an aqueous solution: moles/L
o
Pure solids
(crystals) that are not used as solutes do not have a composition unit.
·
Specify a
temperature in the range 200K to 5000K (298.15K is the default temperature).
·
Optionally specify a
pressure factor of greater than or less than 1 to indicate increasing/decreasing
pressure after the reaction reaches equilibrium at some constant pressure (the
default is a pressure factor of 1, indicating no change to the pressure).
Set the
"Pressure Factor" to a value between 0.1 and 10. This option is effective only
when all reactants and products are gas species (or excluded liquids/solids),
with unequal numbers of reactant and product molecules.
·
Click “Run the
Reaction” to perform the equilibrium thermodynamic calculations and simulate
the reaction until a final steady state is reached. Results are provided in
tabular and graphical forms.
·
See examples 1-15
below. In addition, examples 23-28 include scenarios where the pressure factor
is set greater than or less than 1 (to increase or decrease the pressure,
relative to the baseline pressure, after reaching equilibrium under a
constant baseline pressure) for selected gas-phase reactions.
·
The results panel at
the bottom of the page shows the final compositions with and without pressure
adjustment. The chart shows the reaction progress and kinetics without the
pressure adjustment (since the pressure adjustment is applied only after
reaching equilibrium).
2. With "Select Ionization Reaction"
checked
(for the aqueous ionization/dissociation reactions of
buffers and other compounds):
·
Select a
buffer/compound (including 1st, 2nd or 3rd ionization) from the single dropdown
list below the checkbox. The dropdown list is searchable, so you can type a
partial buffer/compound name and find all matching items. This will populate the
reactant and products automatically.
·
Enter the initial
composition for the reactant and products in moles/L
·
Specify a
temperature in the range 200K to 5000K (298.15K is the default temperature).
· (The
pressure factor will have no effect since none of the reactants or products is a
gas.)
·
Click “Run the
Reaction” to perform the equilibrium thermodynamic calculations and simulate
the reaction until a final steady state is reached. Results are provided in
tabular and graphical forms.
·
See examples 16-17
below.
3. With either of the two reaction modes described above, you can check
"Specify Reaction Rate Parameters" and enter parameters to model
the reaction kinetics during the simulation:
·
ChemReaX supports this generalized reaction rate model: Rate = k *
[R1]X
* [R2]Y * [R3]Z, where R1, R2 and R3 are the reactants.
The constants k, X, Y and Z are experimentally determined for a given reaction.
·
Enter the constants
k, X, Y and Z into the text boxes -- these constants must be in the correct
units (which are not checked by ChemReaX). Note that the reactant concentrations
are in moles/L or bars and time is assumed to be in seconds.
· Instead of entering k, you can provide the A and Ea values for the Arrhenius equation as an alternative (if A is a large number, specify it using scientific notation -- for example, 7.5e13). ChemReaX will then calculate the reaction rate k as a function of temperature.
·
See examples 18-22
below which illustrate a single reaction modeled as zeroth order through third
order. The chart shows the reaction progress and kinetics based on the reaction
rate model provided by the user. In addition, ChemReaX provides detailed data
for the reactant/product compositions over time in tabular form based on the
kinetics.
4. Additional options:
· Click "Get
Thermodynamic Properties"
to get only the standard molar
enthalpy of formation and the molar entropy for each of the reactants and
products at the specified temperature -- typically for convenient look up of
this data for use outside of ChemReaX -- without calculating the thermodynamic
equilibrium values or running the simulation.
· Click "Check
Equation Validity" to verify mass conservation and
charge balance in the given equation. An error is reported if atoms or charges
are out of balance, or if the given reactants cannot produce the required
products. This check is also done automatically when "Run the Reaction"
is clicked.
· Click "Balance
the Equation" to automatically perform a stoichiometric
balancing of the equation. You can clear the coefficients by clicking "Clear
Balancing".
Examples of Setting Up and Simulating General Reactions Using ChemReaX
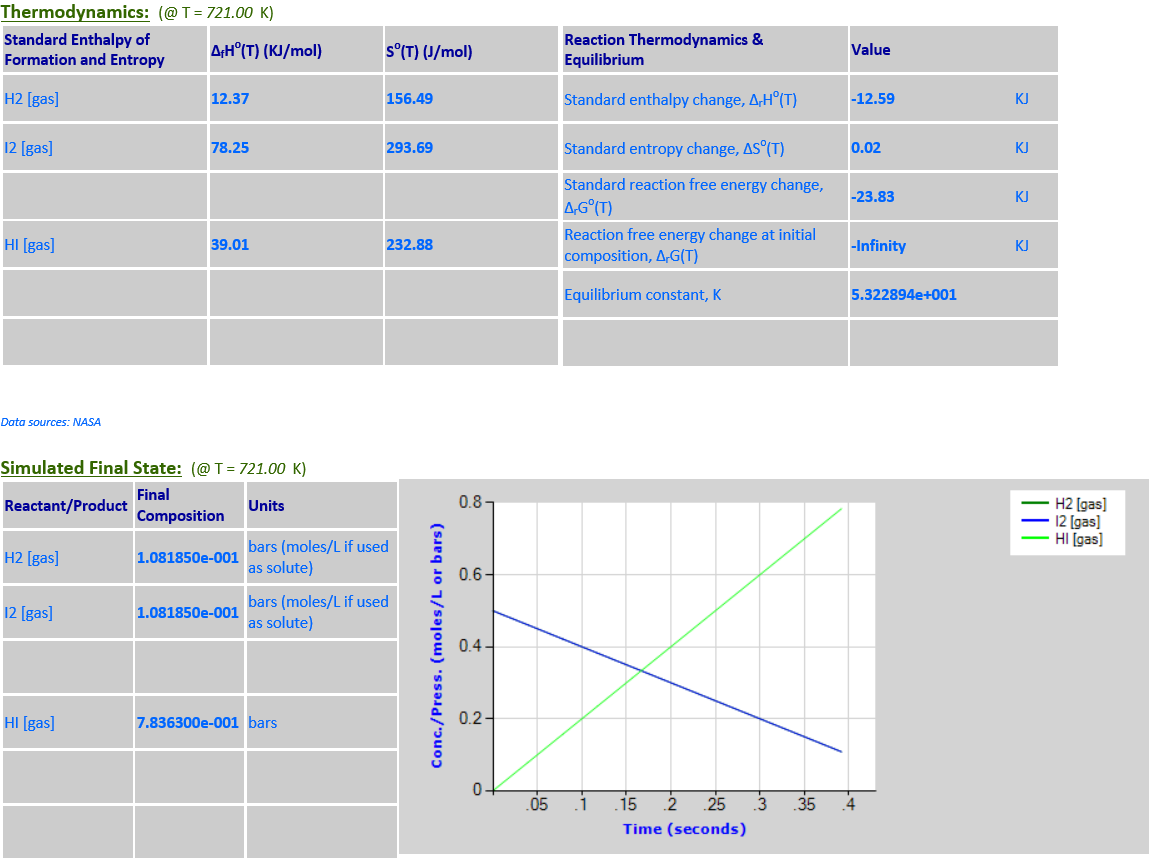
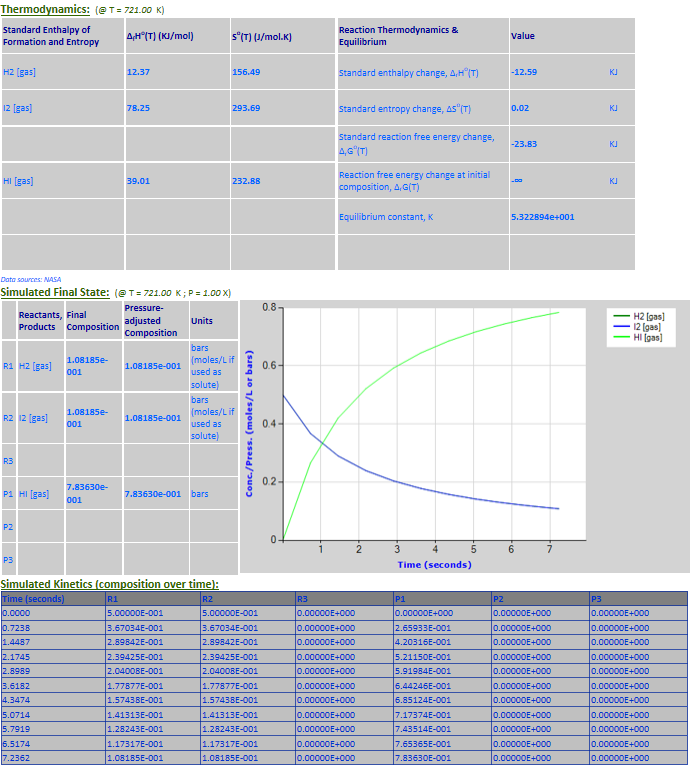
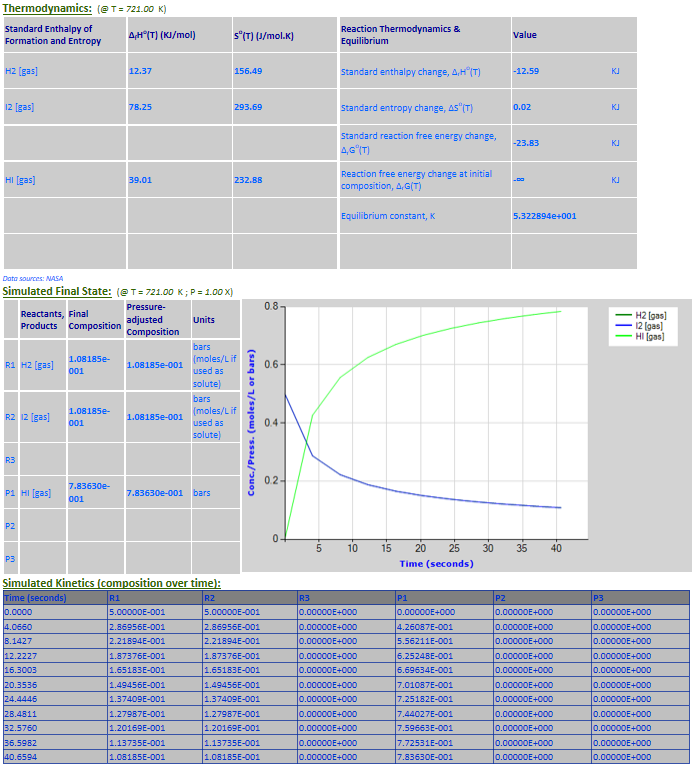
1.
H2 (g) + I2 (g) <--> 2HI (g)
·
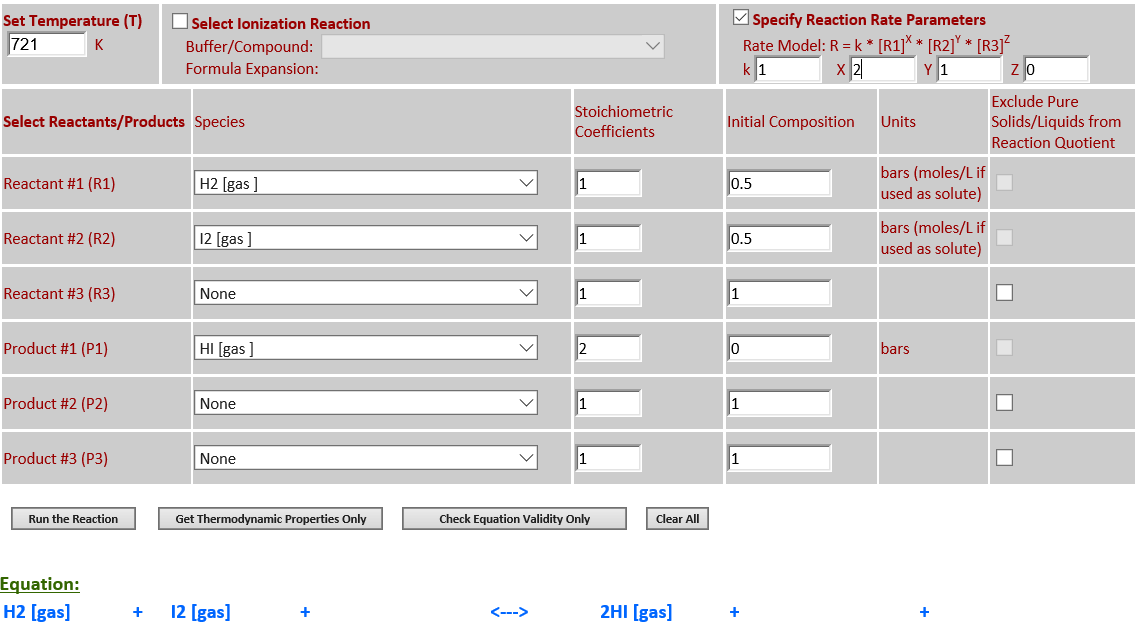
·
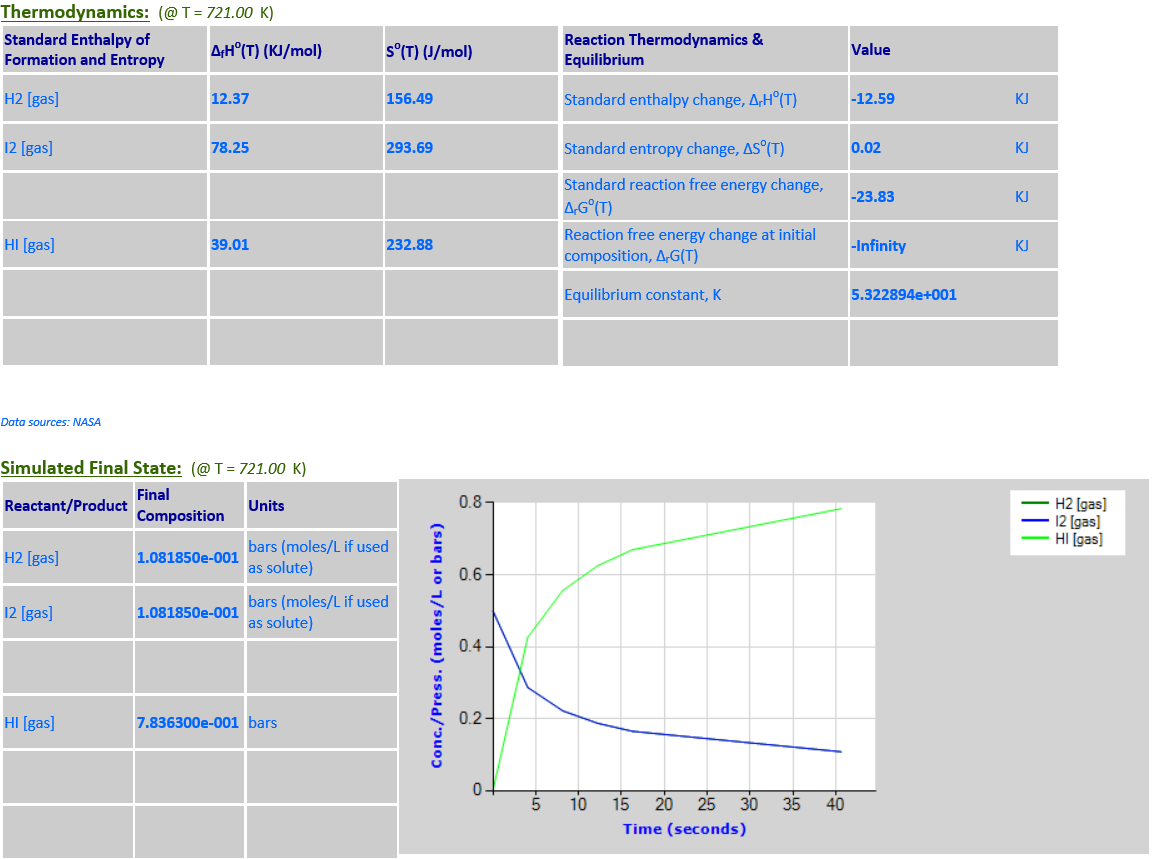
2.
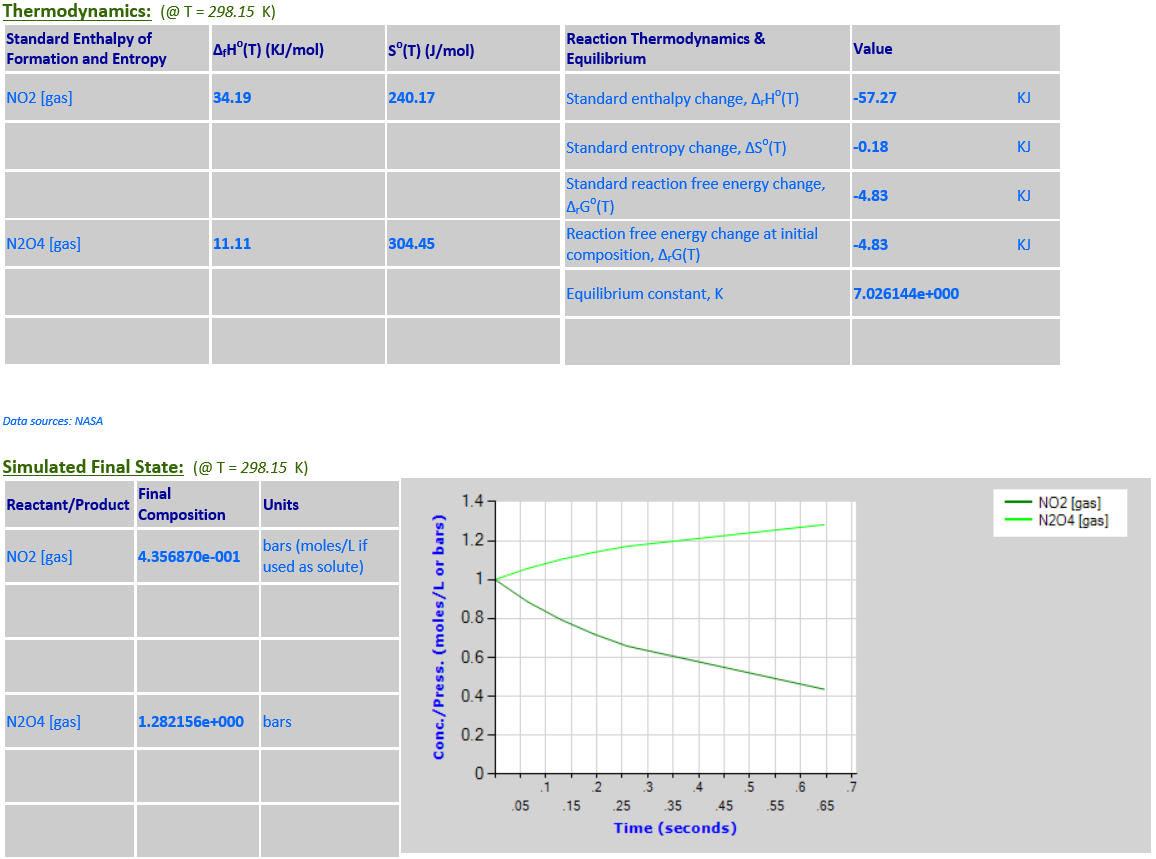
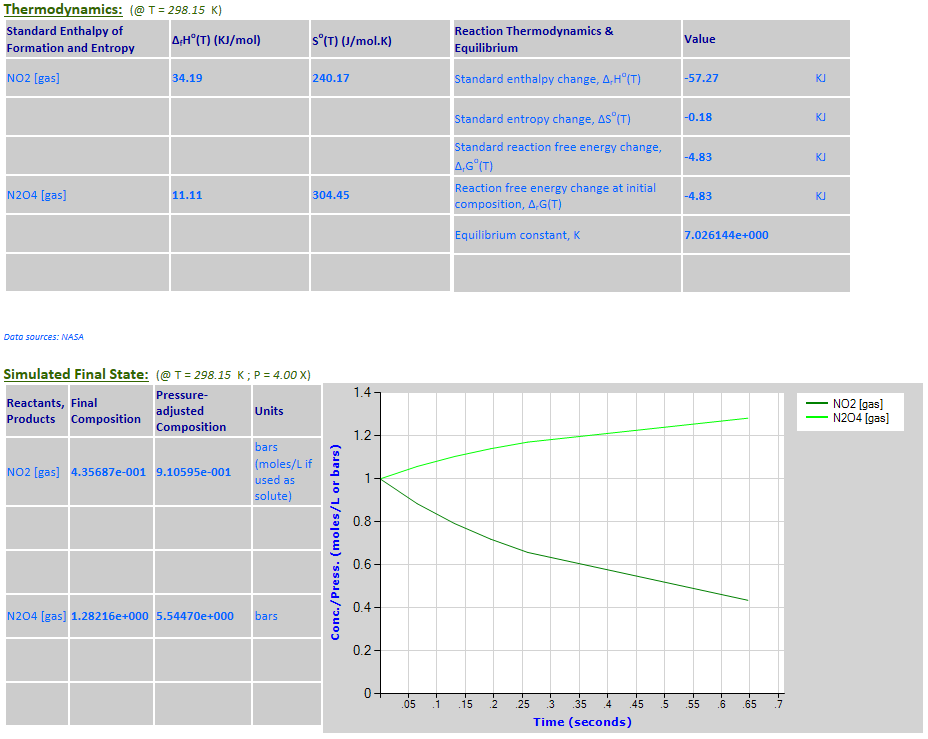
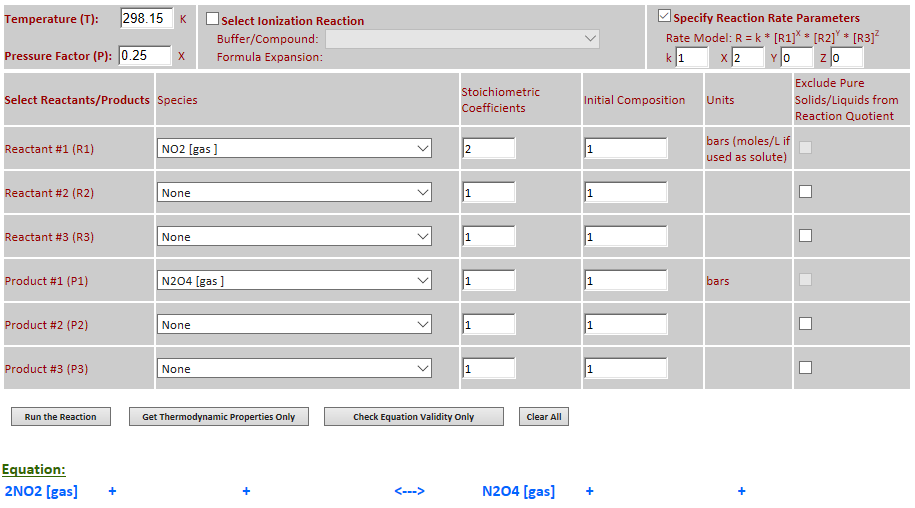
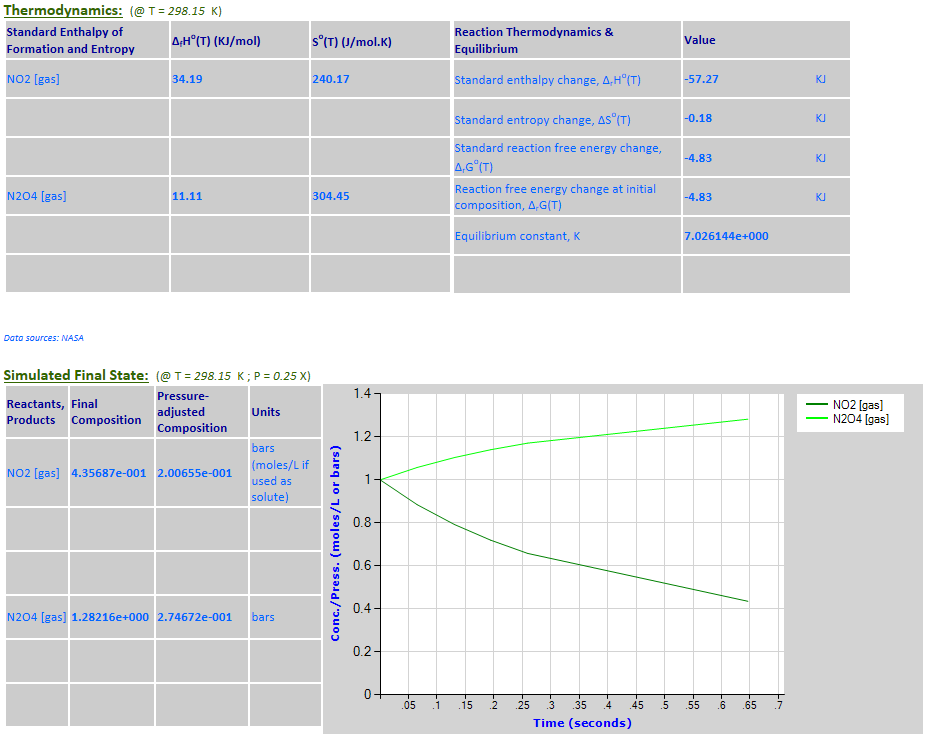
2NO2 (g) <--> N2O4 (g)
·
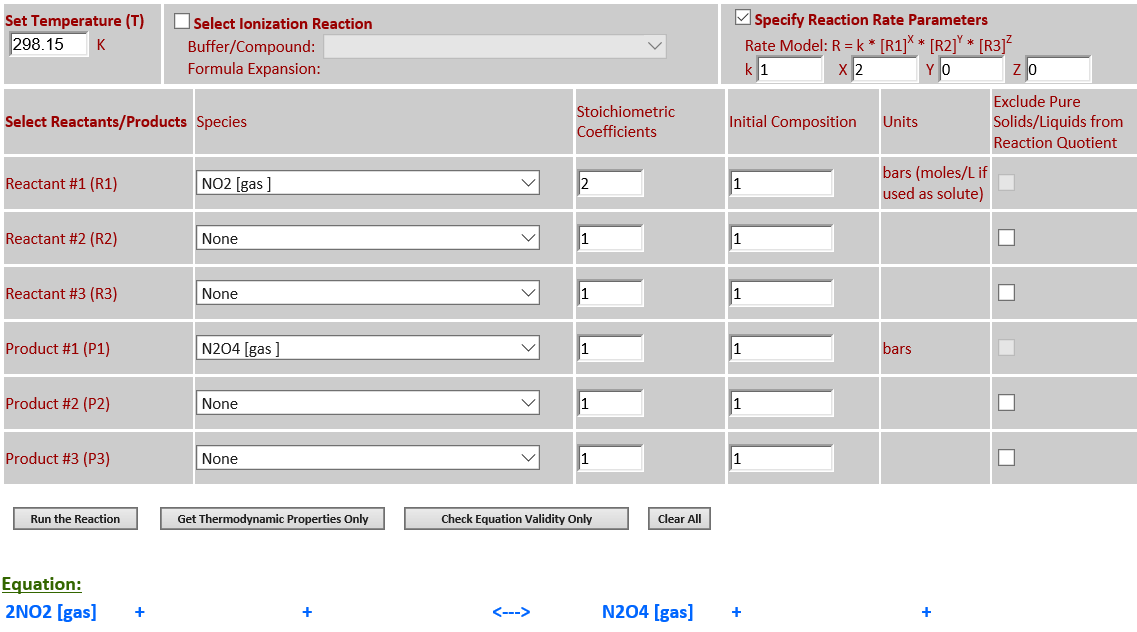
·
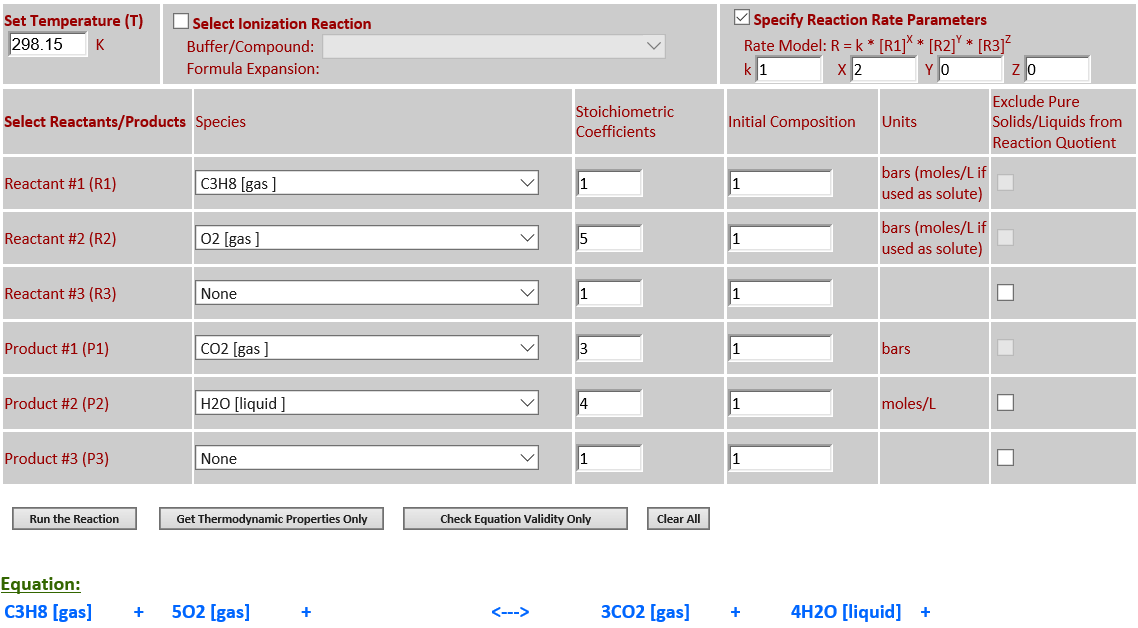
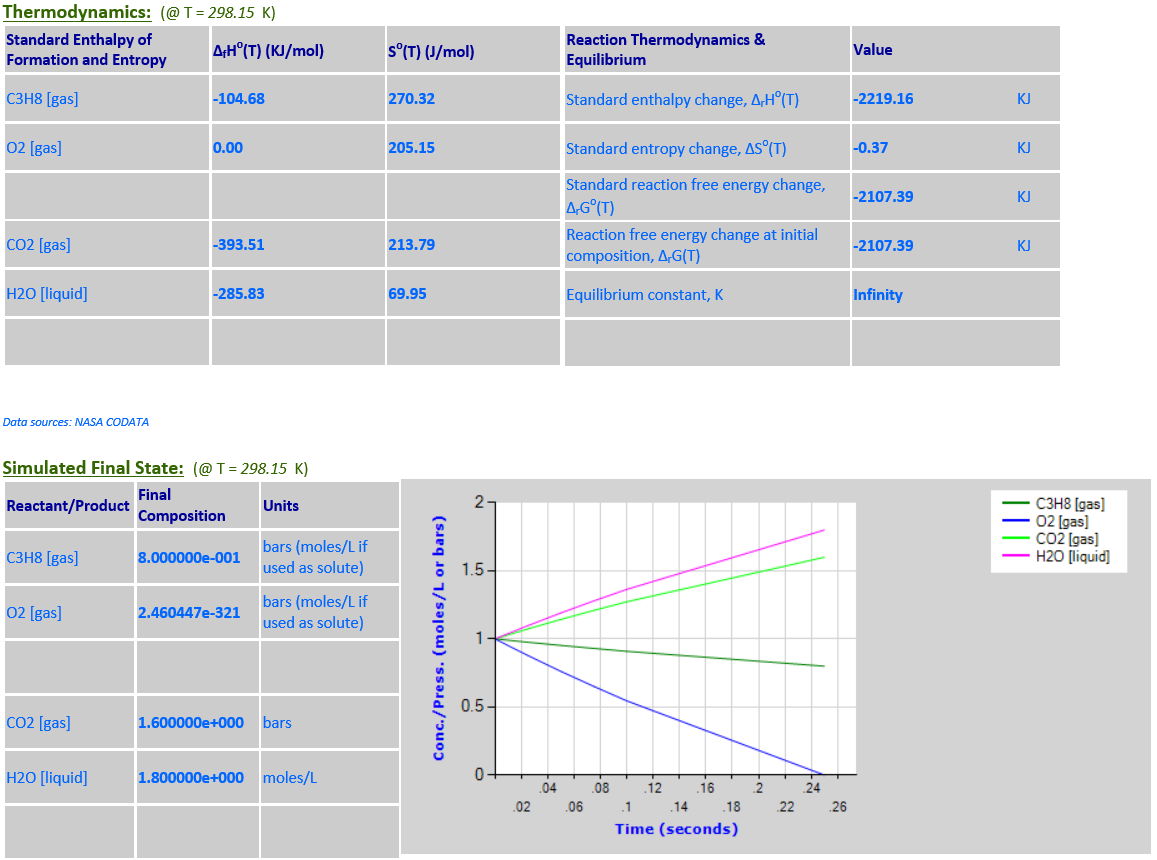
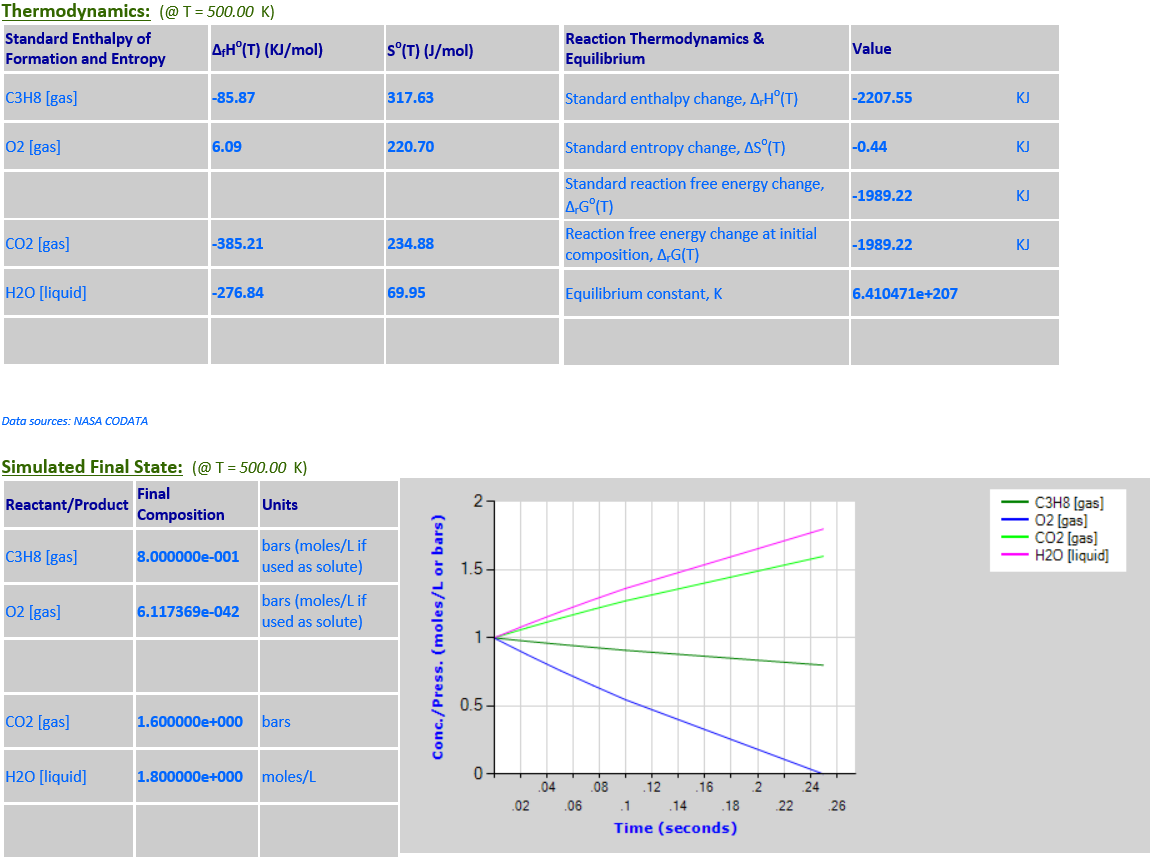
3.
C3H8 (g) + 5O2 (g) <--> 3CO2 (g)
+ 4H2O (l)
·
O2 is a limiting reactant. H2O (liquid) is a product that must be
retained in the reaction quotient since it is not a solvent and its
concentration is comparable to other reaction components. As the temperature
increases from 298.15K to 500K, the equlibrium noticeably shifts to the left
towards the reactants in order to absorb some of the heat since
∆rGo(T)
is negative.
·
·
4.
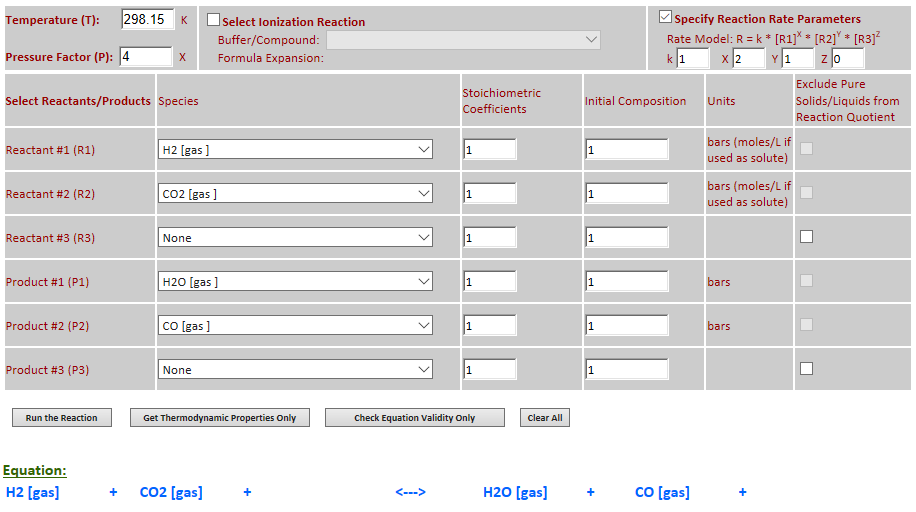
H2 (g) + CO2 (g) <--> H2O (g) +
CO (g)
·
As the temperature
increases from 298.15K to 500K, the equlibrium noticeably shifts to the right
towards the products in order to absorb some of the heat since
∆rGo(T) is
positive.
·
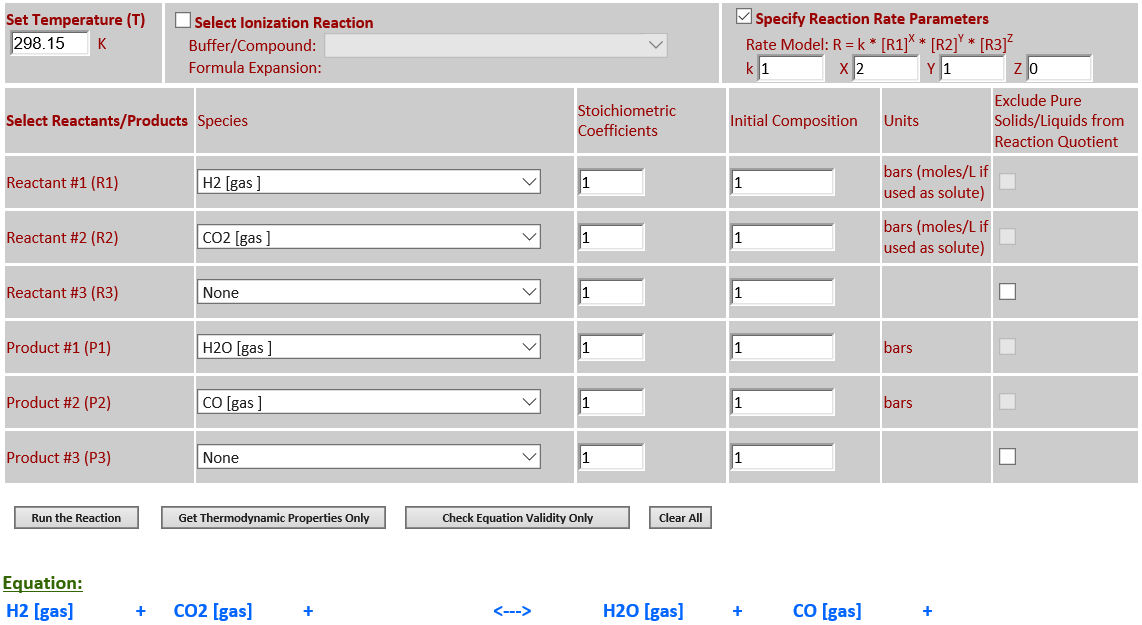
·
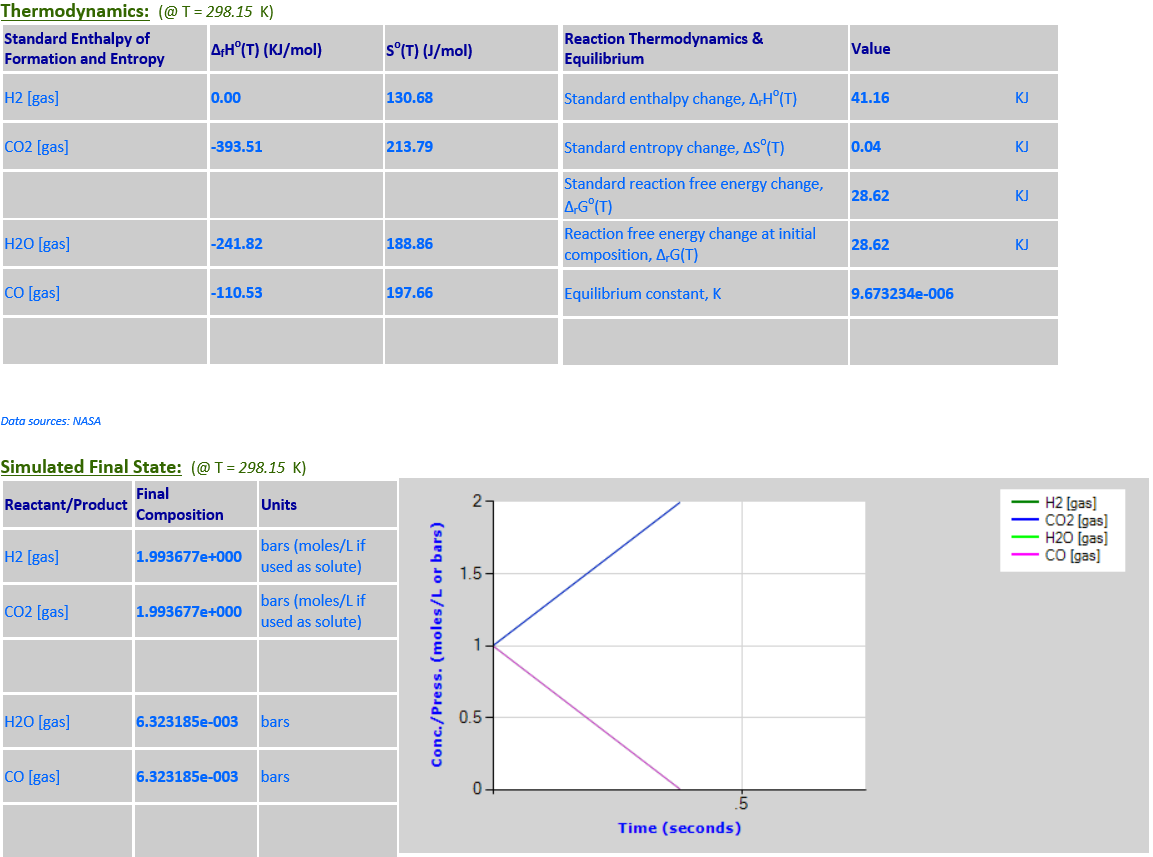
·
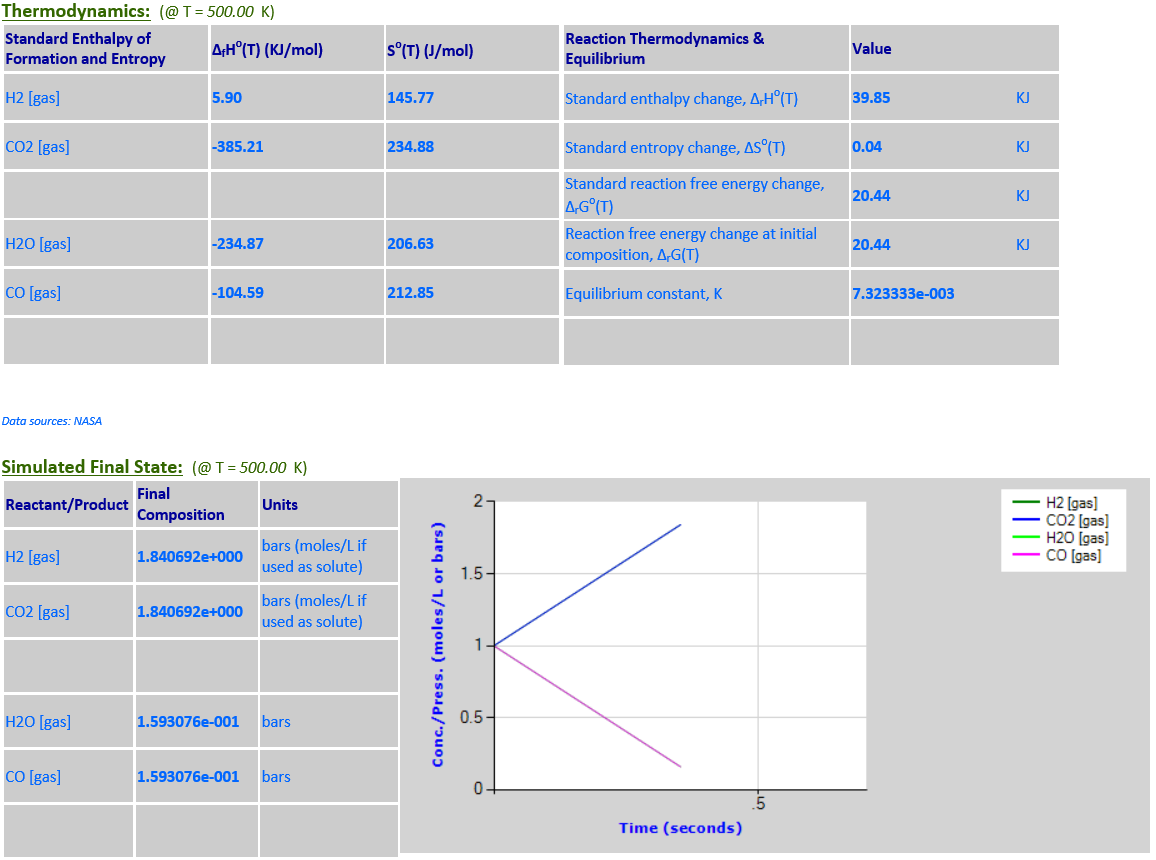
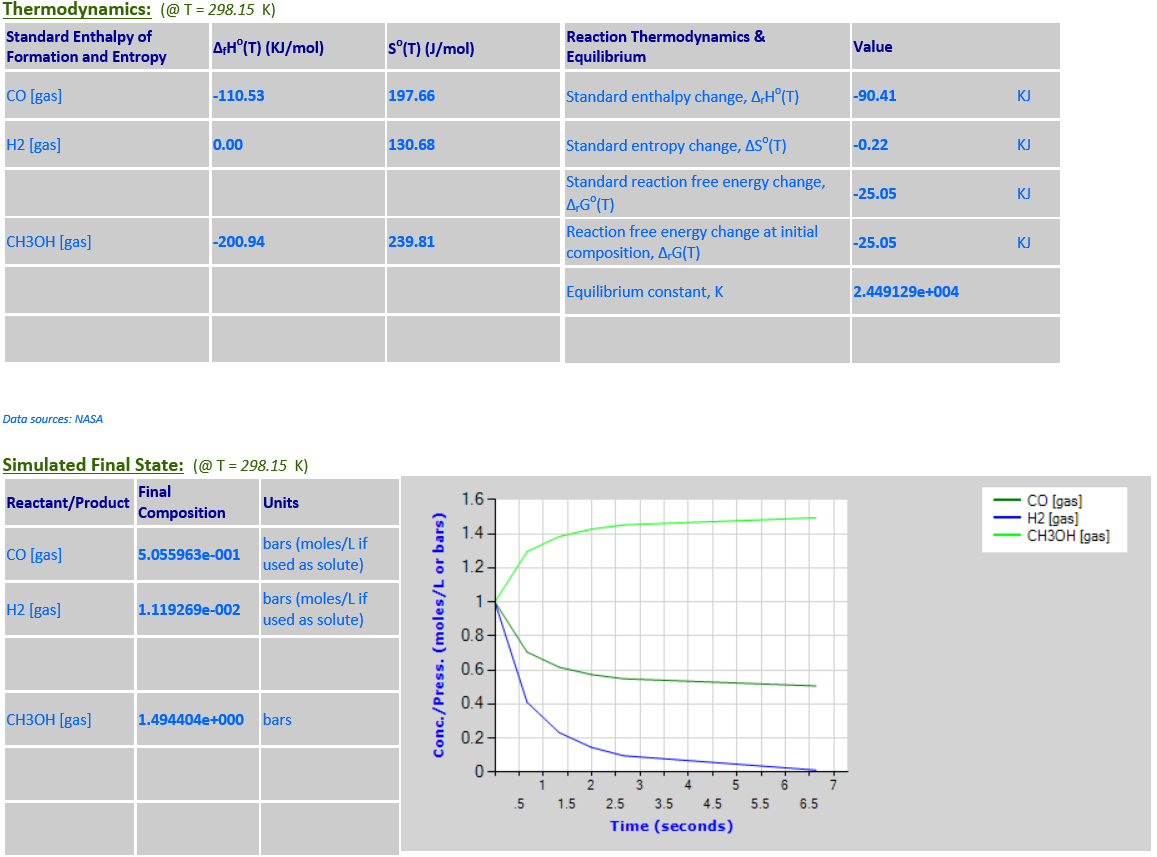
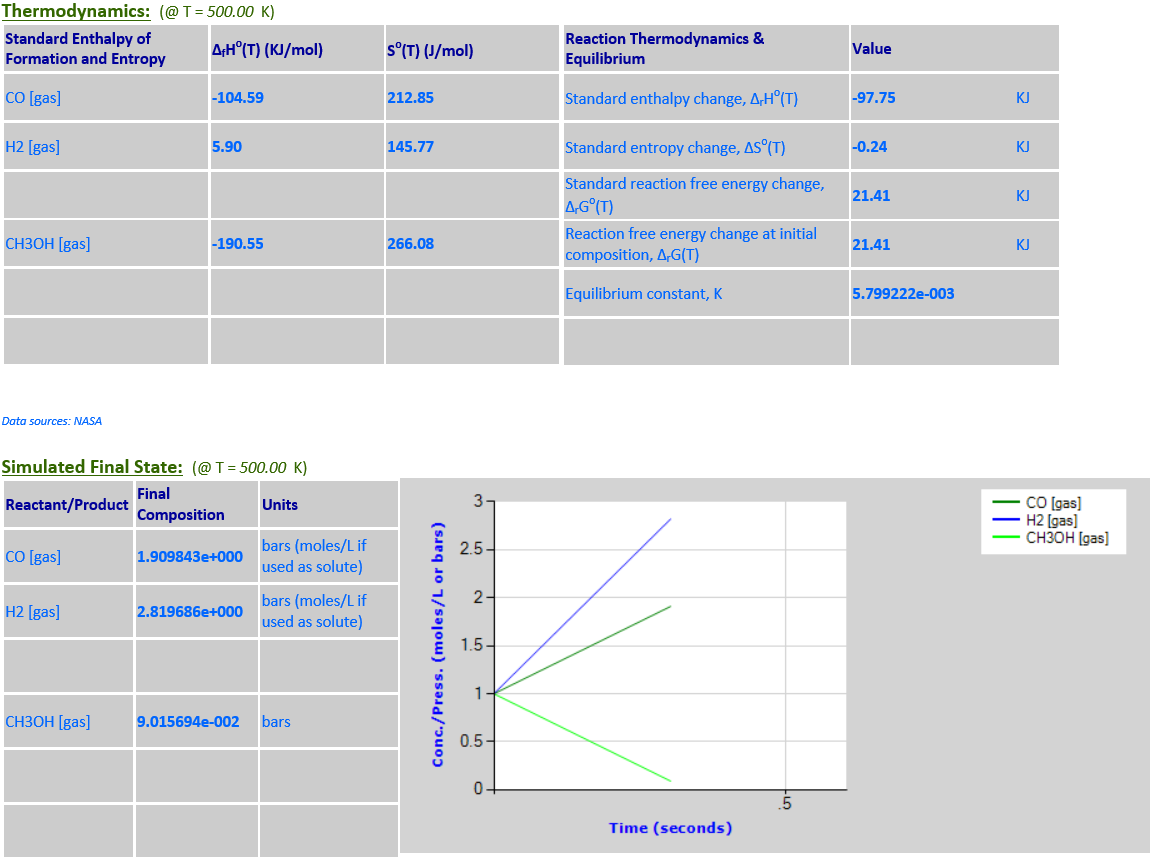
5.
CO (g) + 2H2 (g) <--> CH3OH (g)
·
∆rGo(T) is
negative at 298.15K and the final state is mostly products. As the temperature
increases to 500K,
∆rGo(T) changes sign and becomes
positive, and the final state is mostly reactants.
·
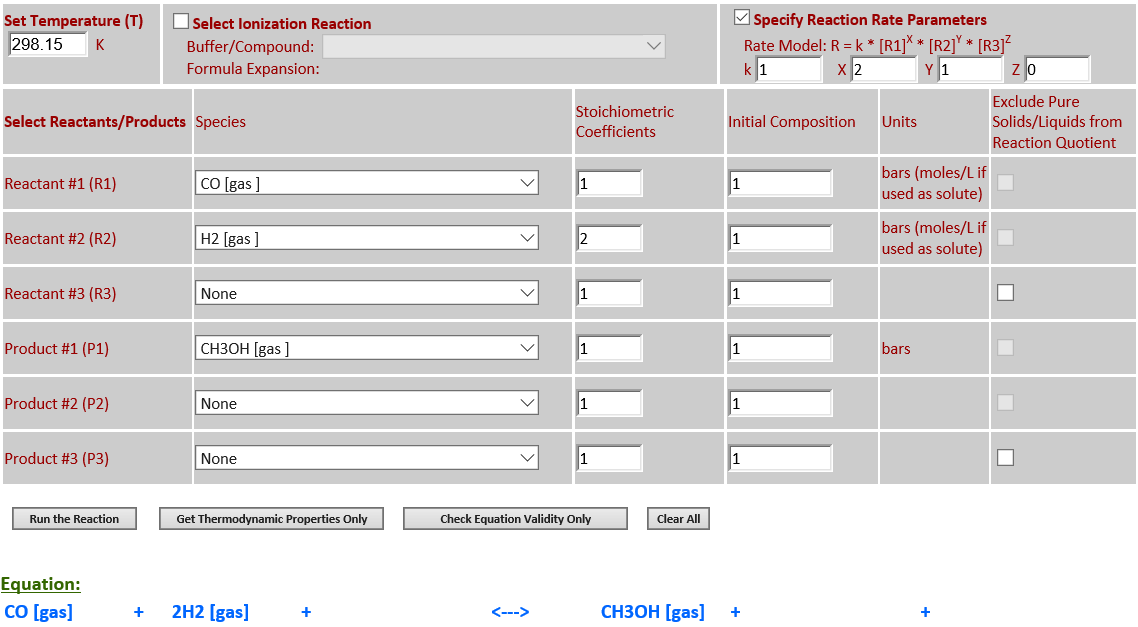
·
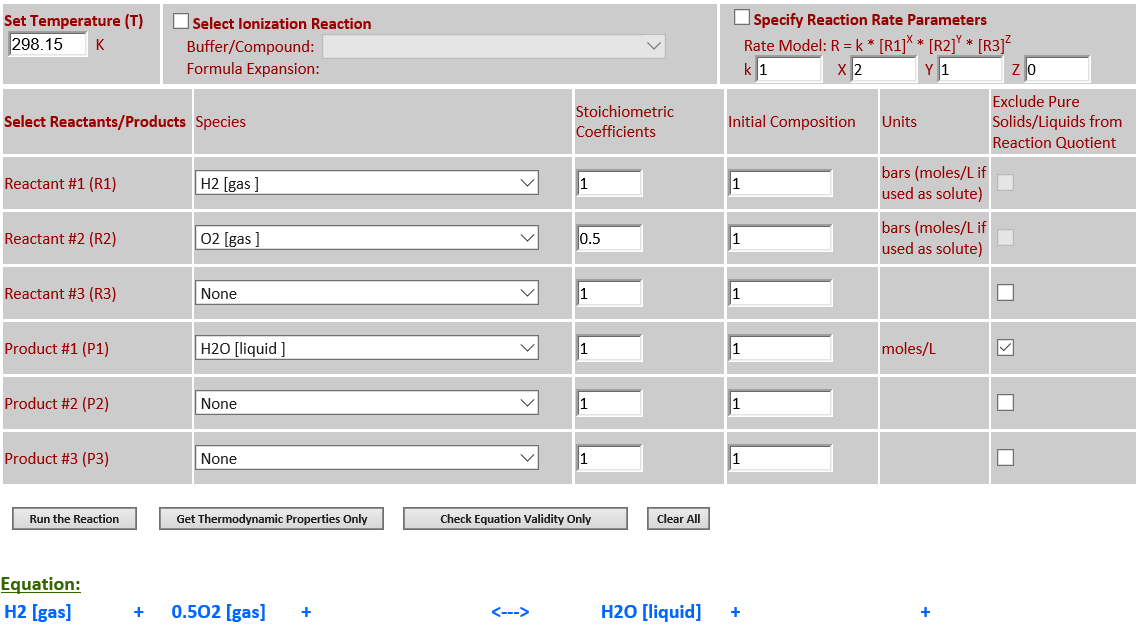
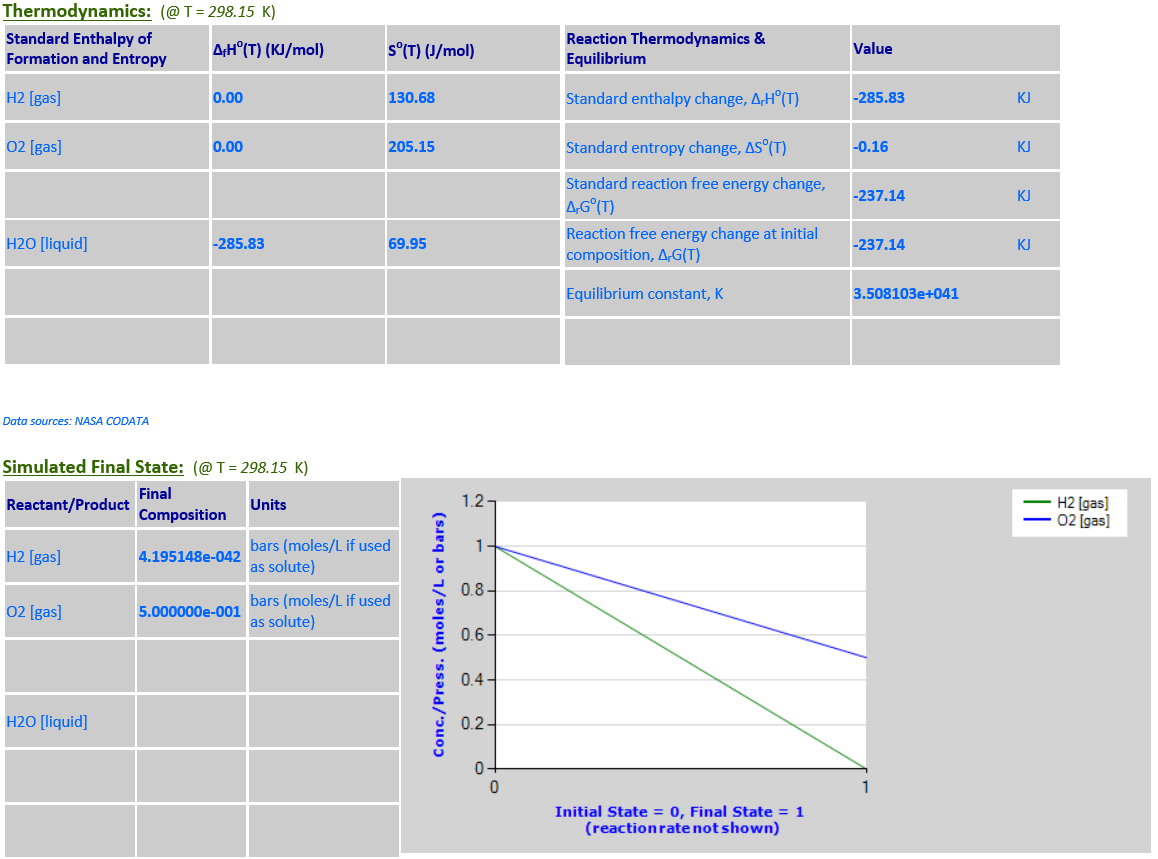
6.
H2 (g) + 0.5O2 (g)
<--> H2O (l)
·
H2O is a pure liquid
that must be excluded from the reaction quotient.
·
·
7.
2Hg (l) + O2 (g) <--> 2HgO (c)
·
Hg is a pure liquid
and HgO is a pure solid – both must be excluded from the reaction quotient
·
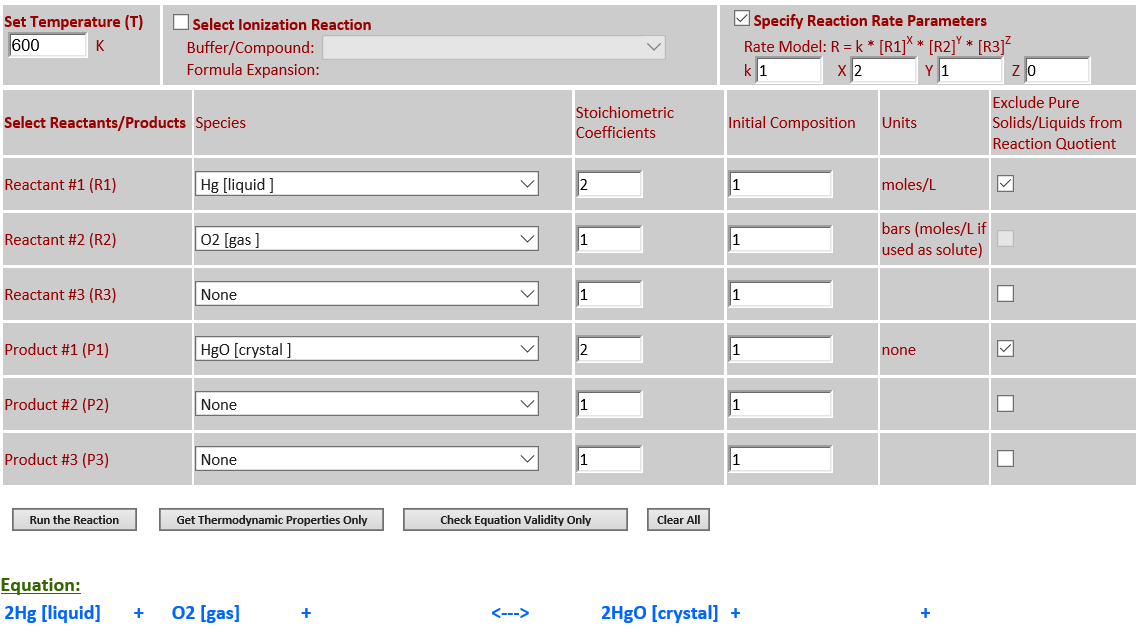
·
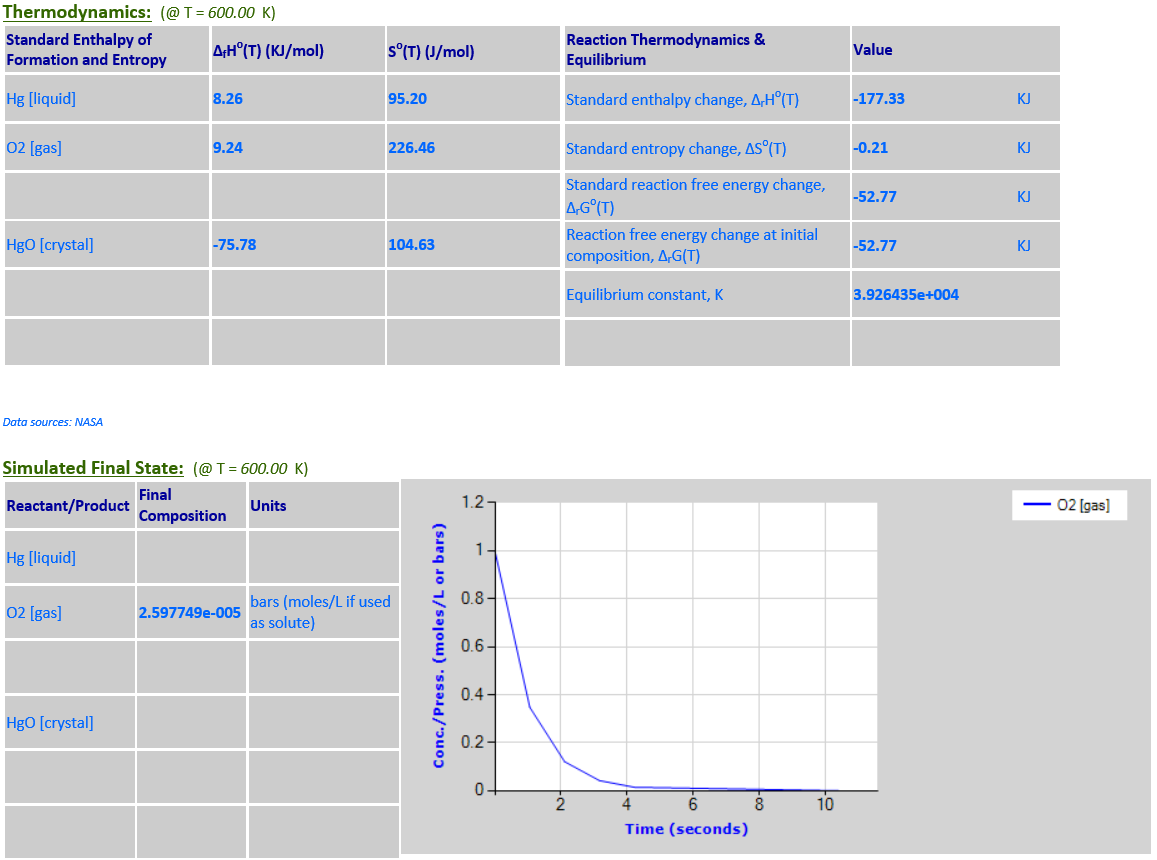
8.
H2O (l) <--> H2O (g)
·
H2O is a pure liquid
that must be excluded from the reaction quotient for the evaporation of water.
No evaporation takes place at 298.15. At 500K, a considerable amount of
evaporation takes place.
·
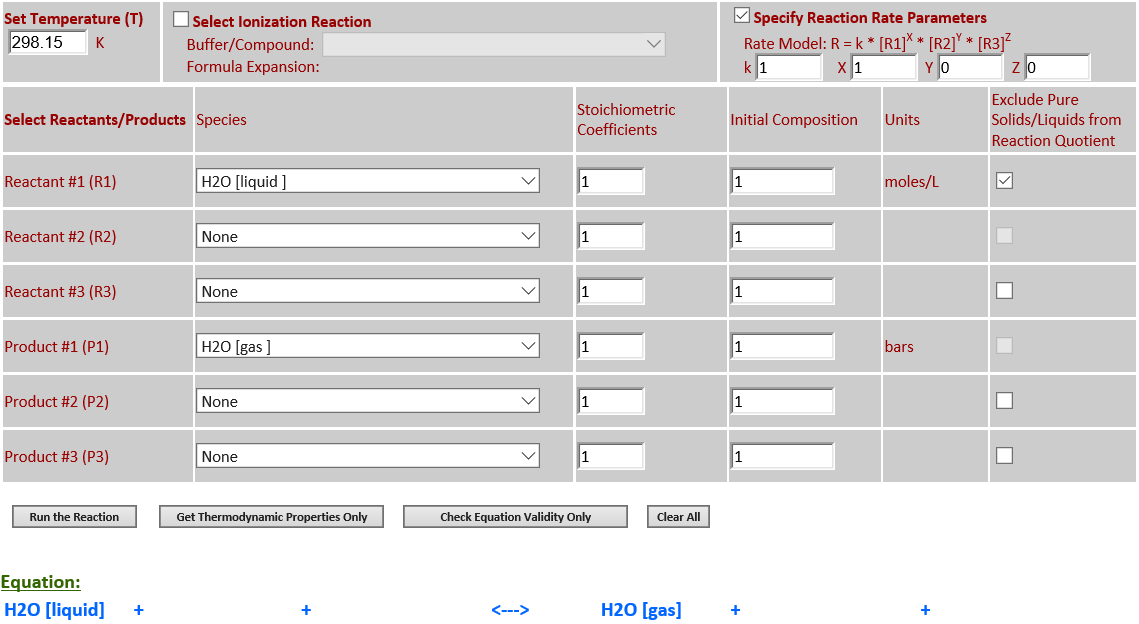
·
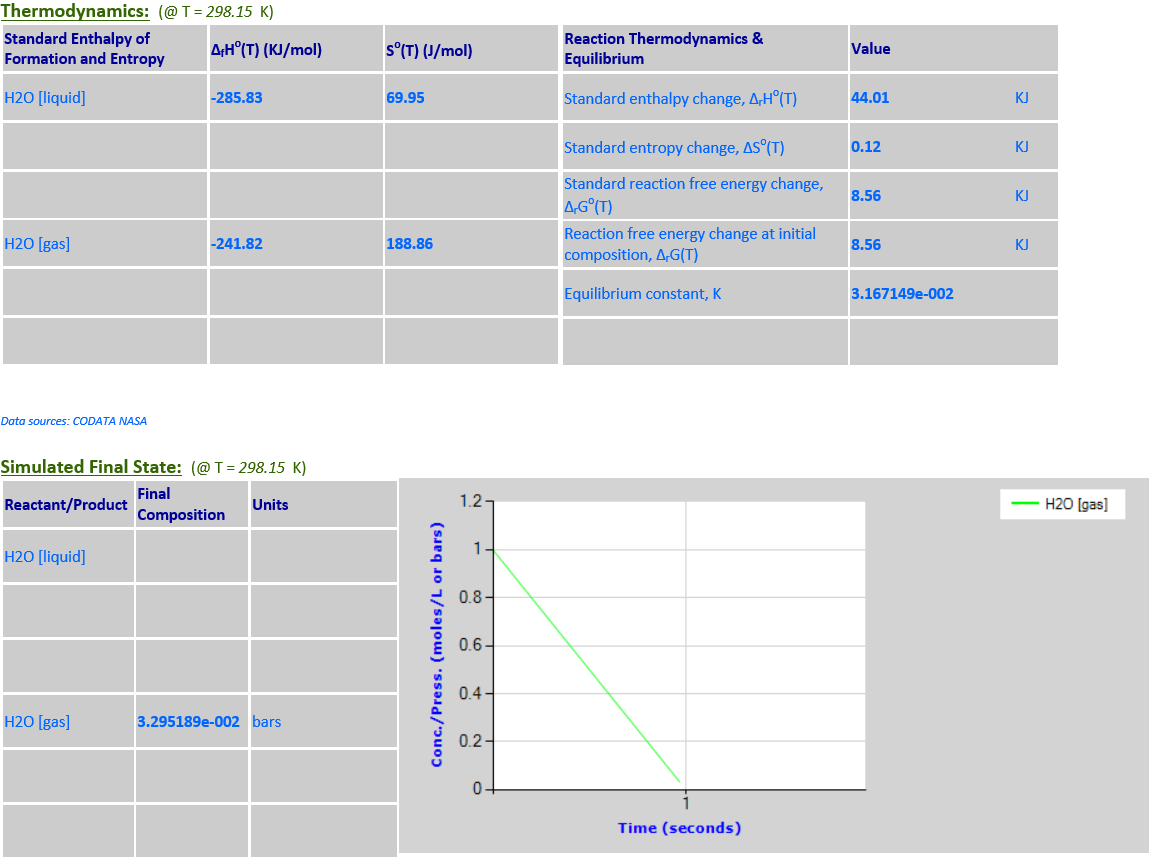
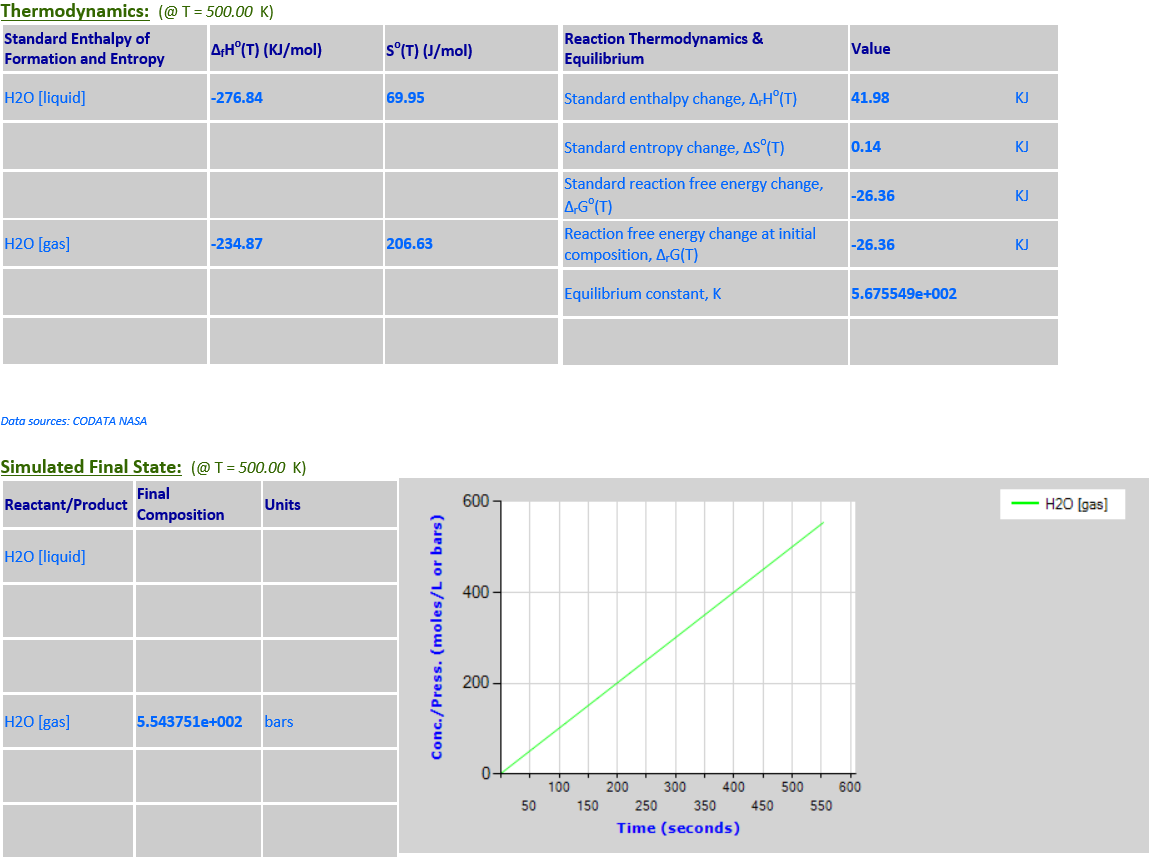
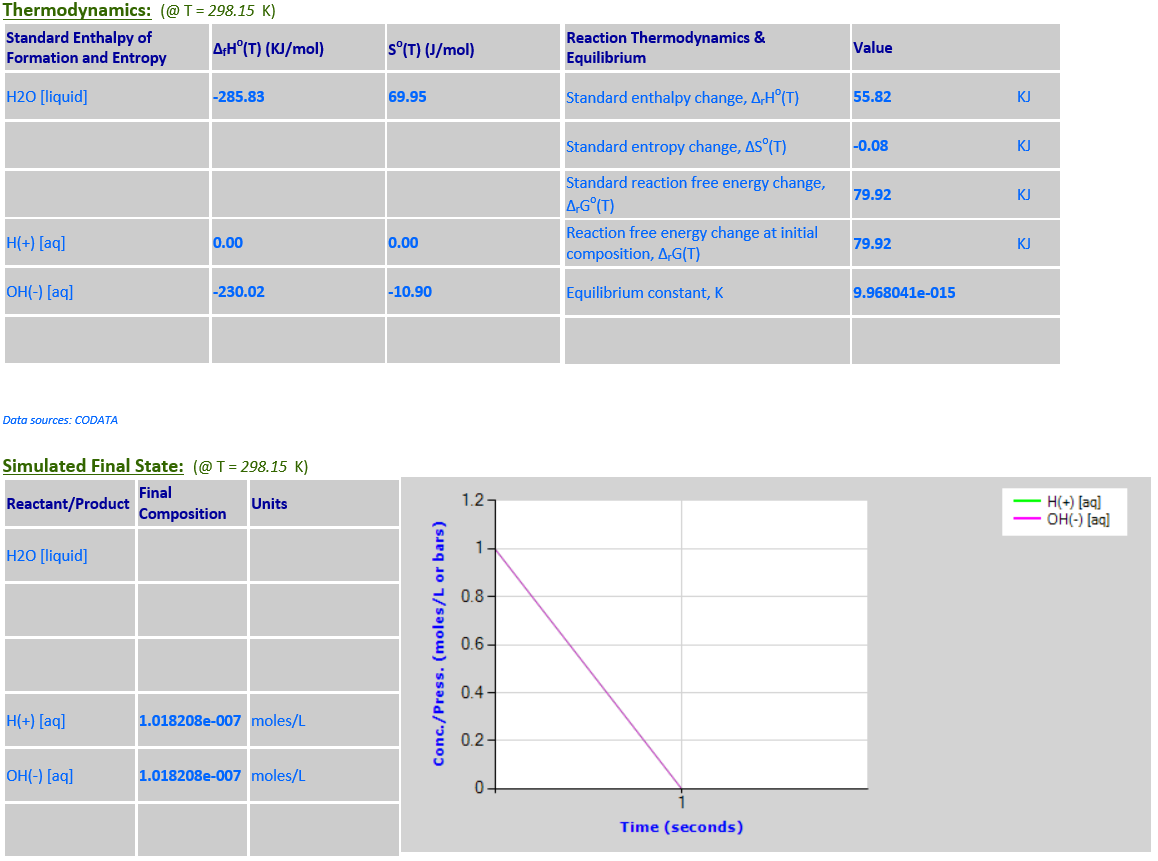
9.
H2O (l) <--> H+ (aq) + OH- (aq)
·
H2O is a pure liquid that must
be excluded from the reaction quotient for the dissociation of water.
·
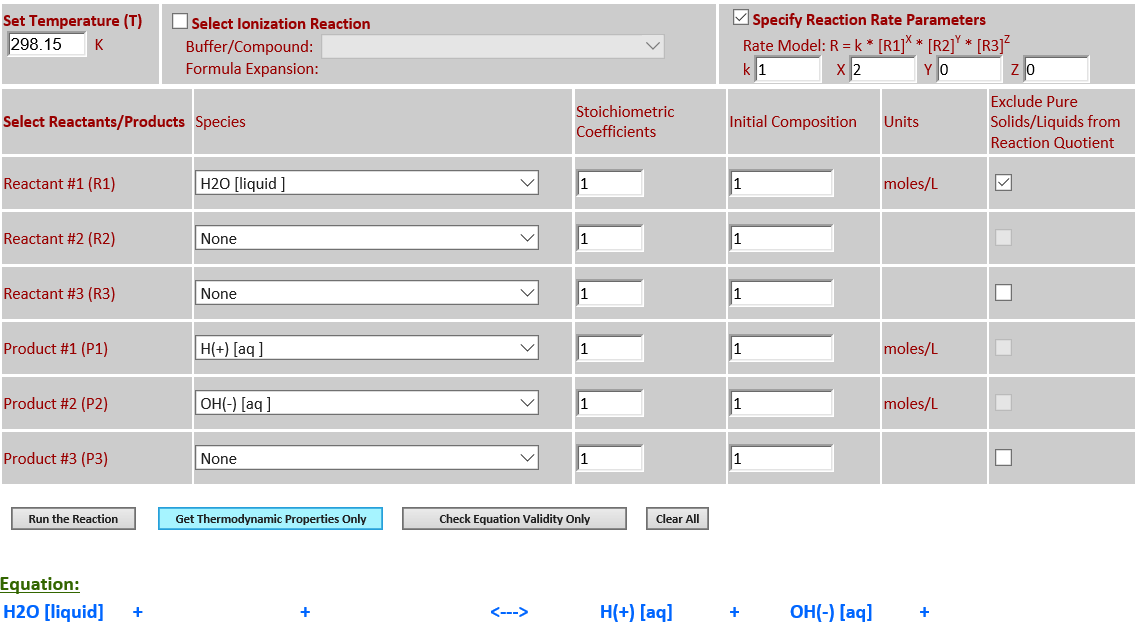
·
10.
2HCl (g) +
F2 (g) <--> 2HF (g) + Cl2 (g)
·
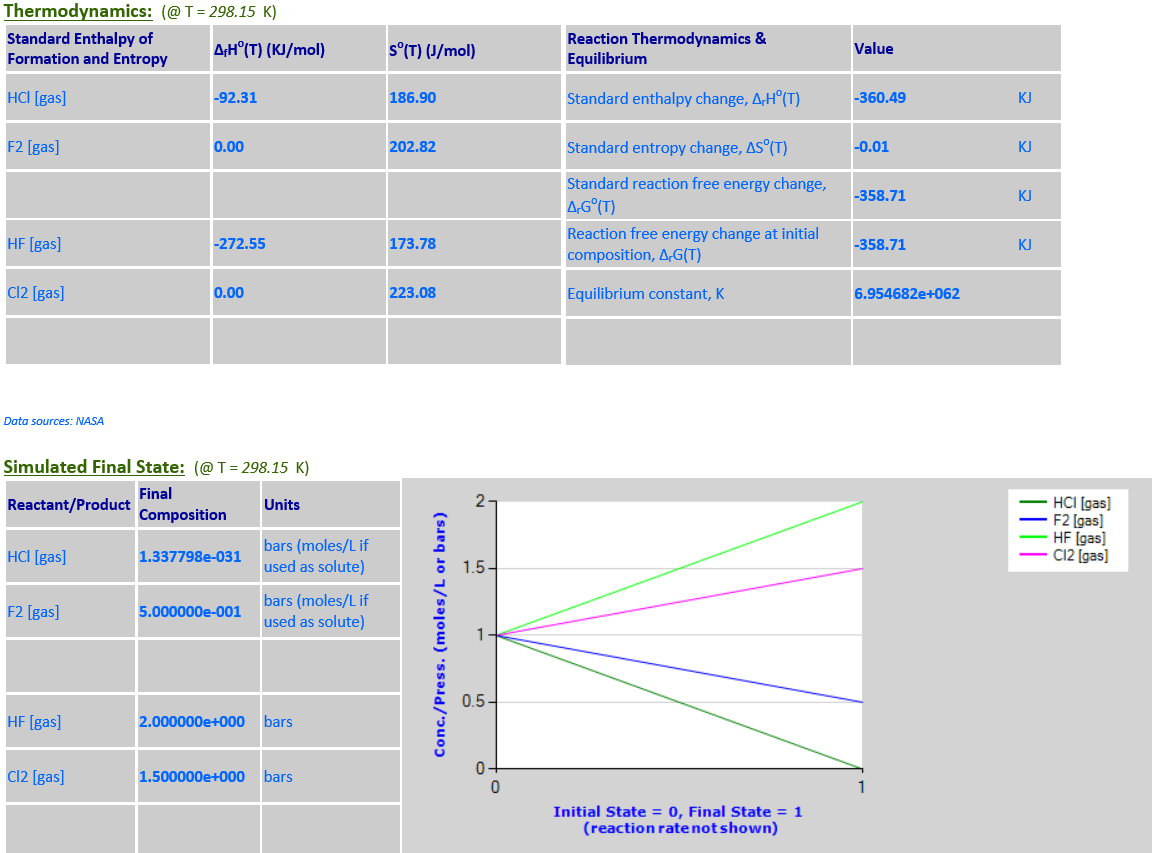
∆rGo(T) is negative
and final state is nearly 100% products.
·
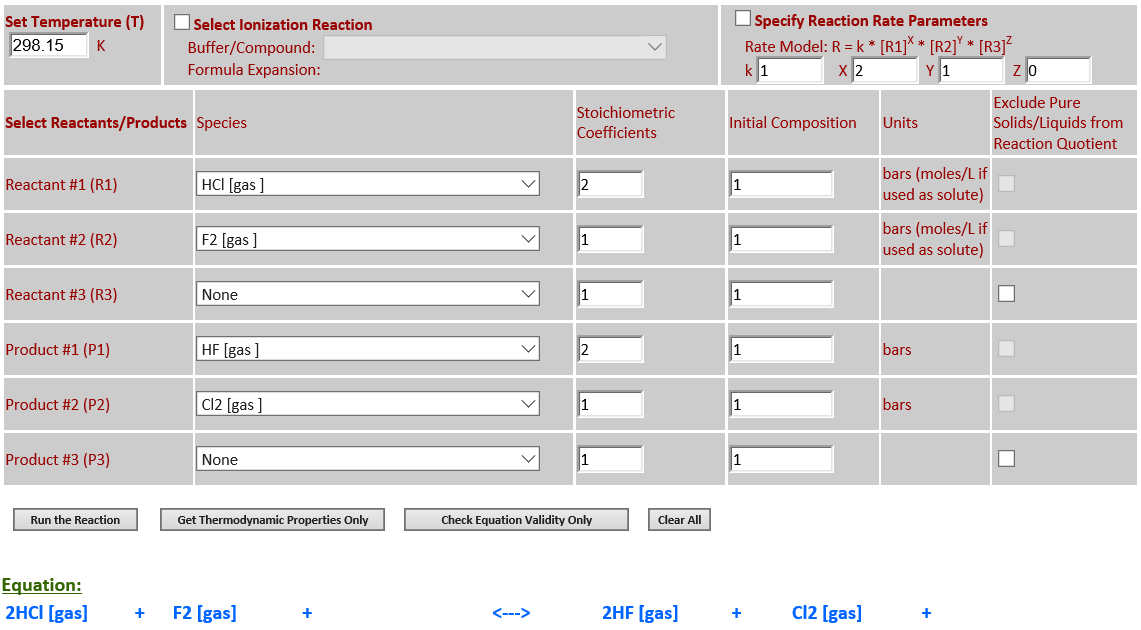
·
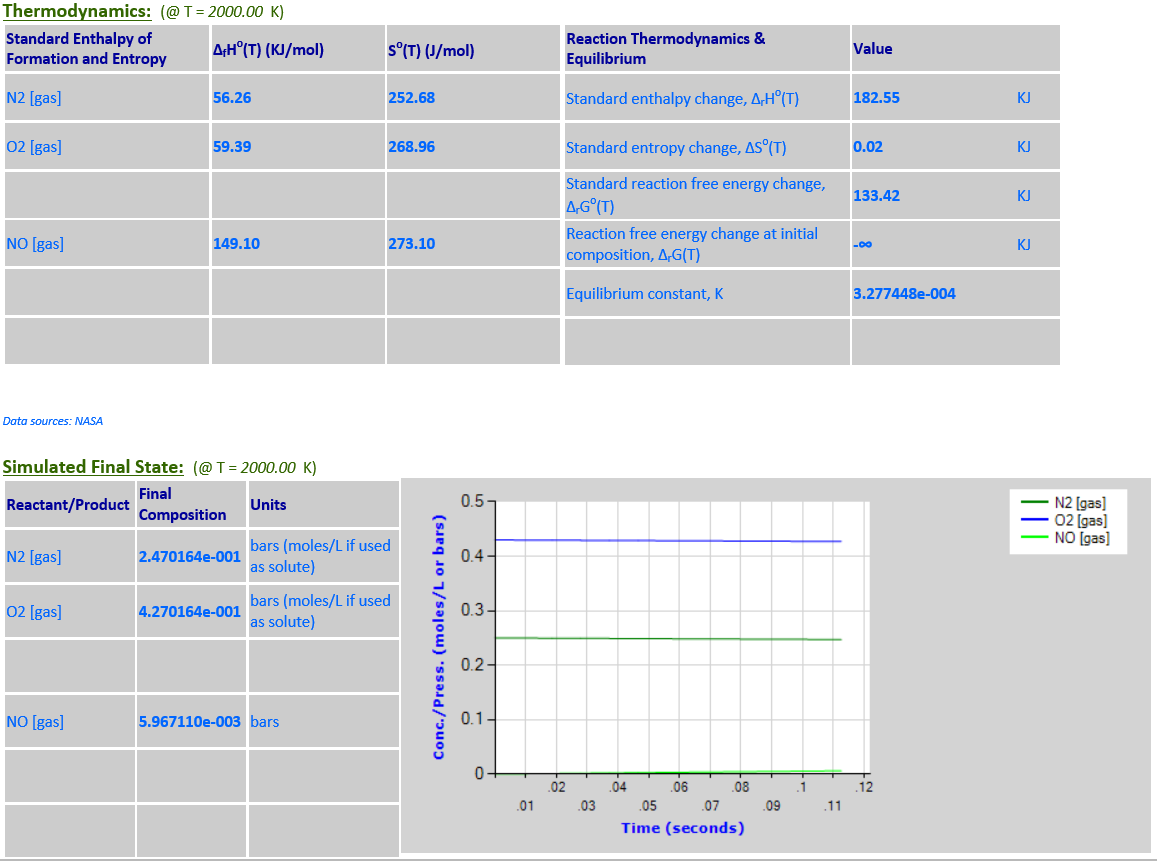
11. N2 (g) + O2
(g) <--> 2NO (g)
·
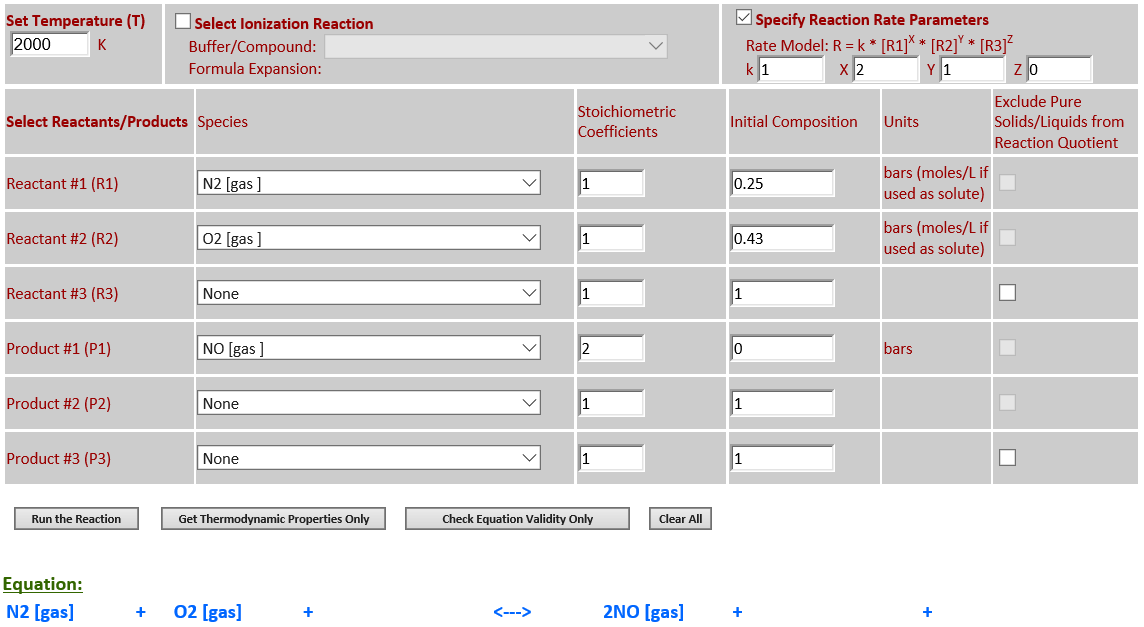
·
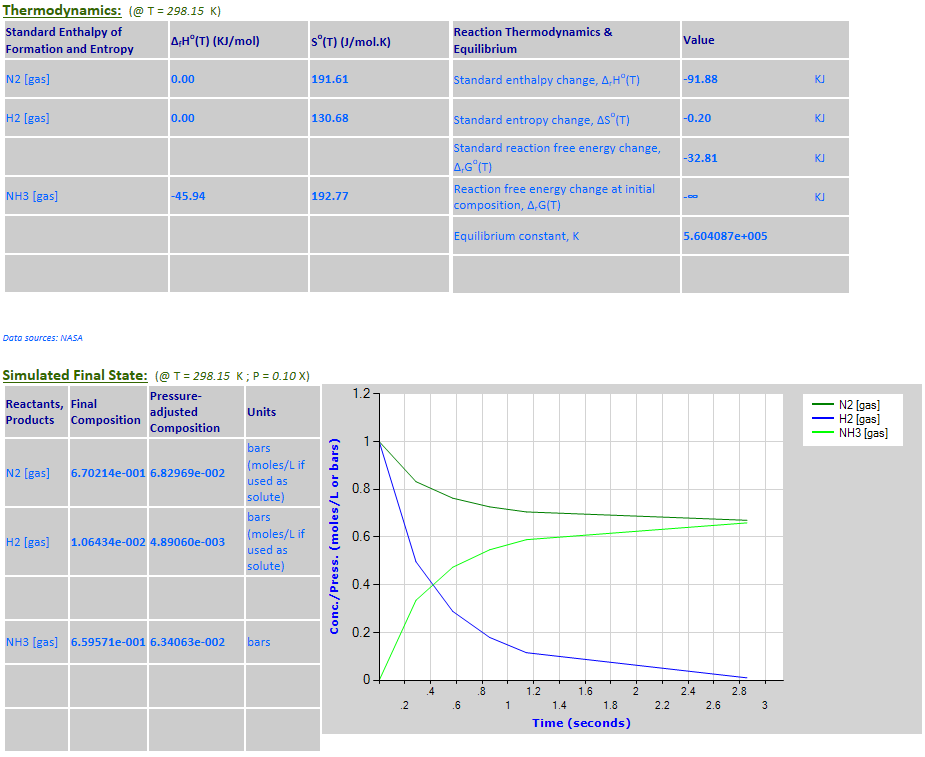
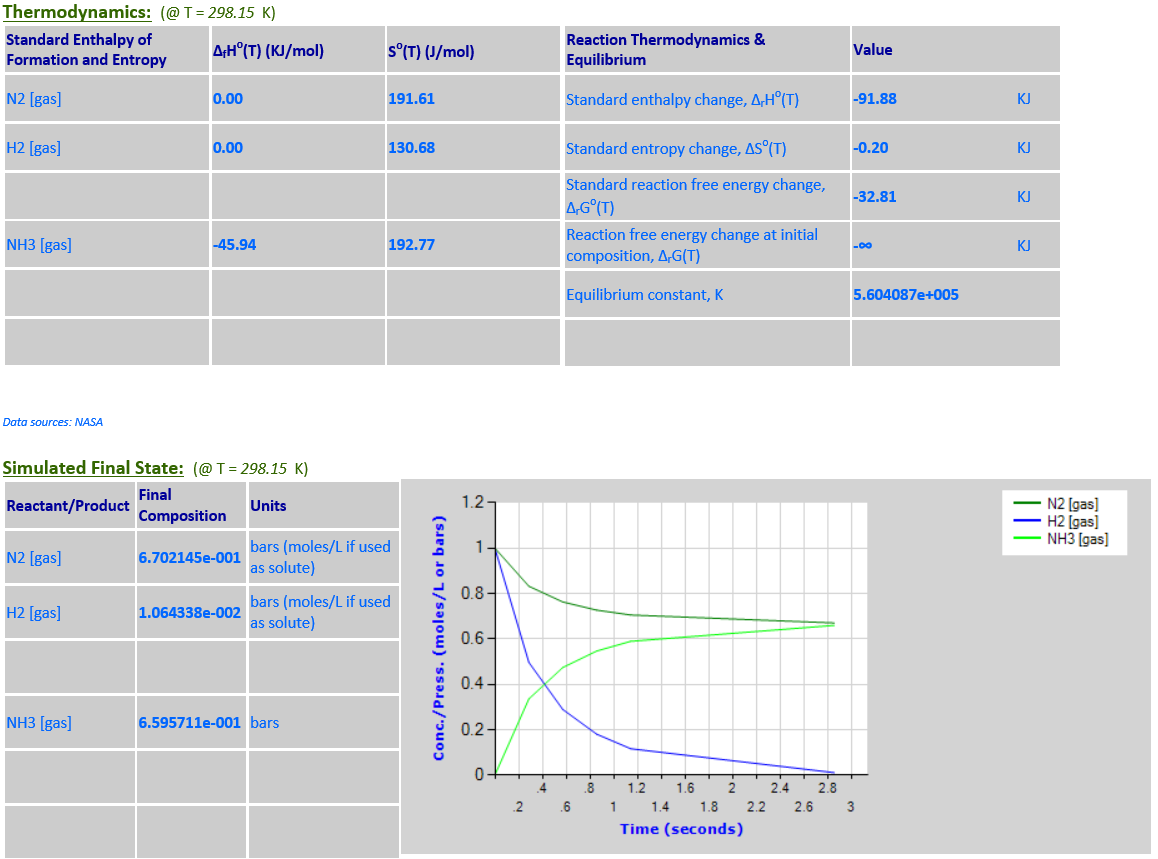
12. N2 (g) + 3H2
(g) <--> 2NH3 (g)
·
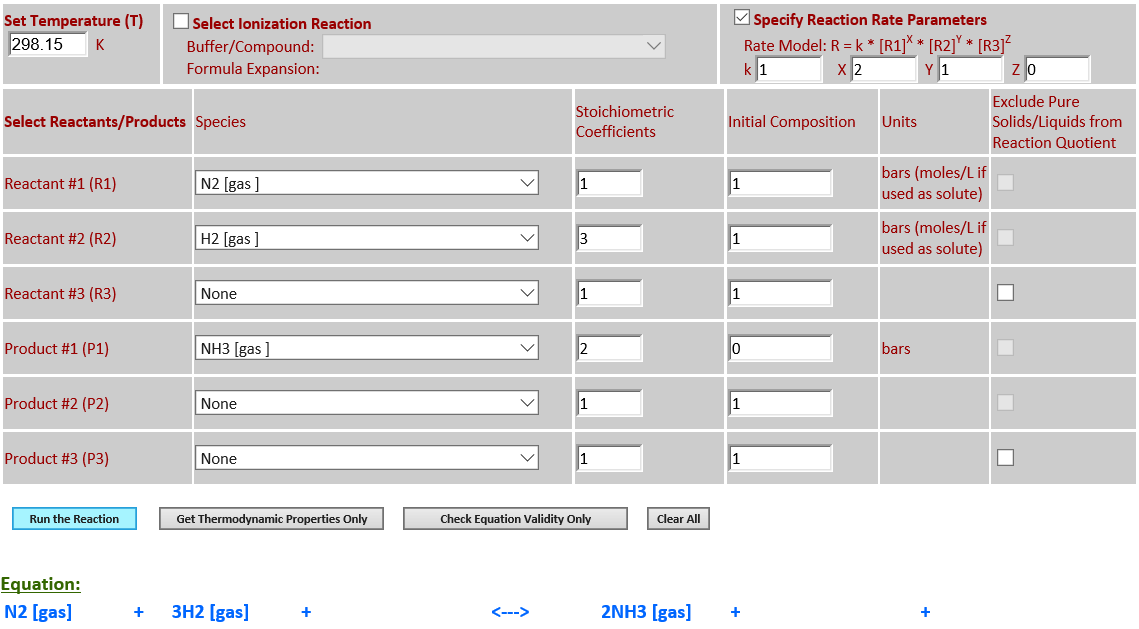
·
13. H2 (g) + Cl2
(g) <--> 2HCl (g)
·
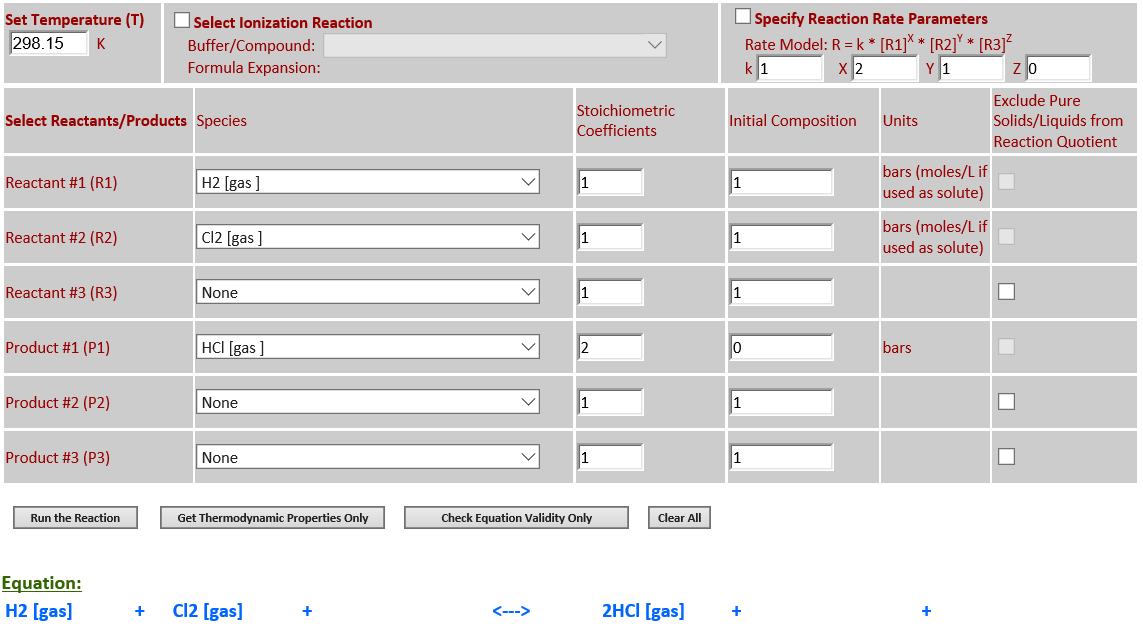
·

14. CH4 (g) + 2O2
(g) <--> CO2 (g) + 2H2O (l)
·
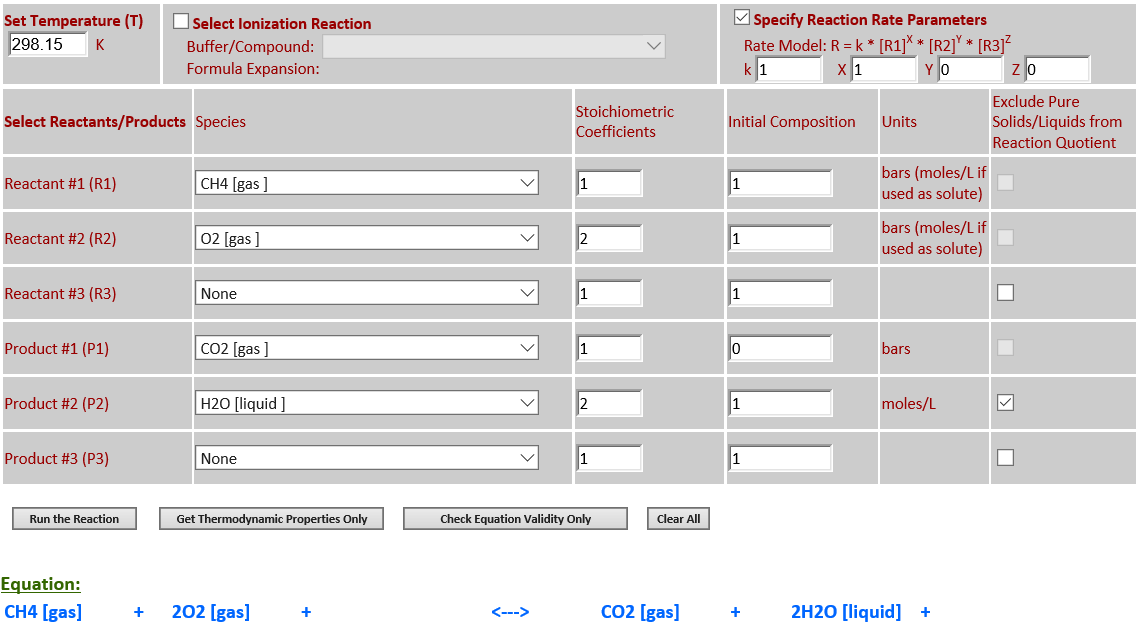
·
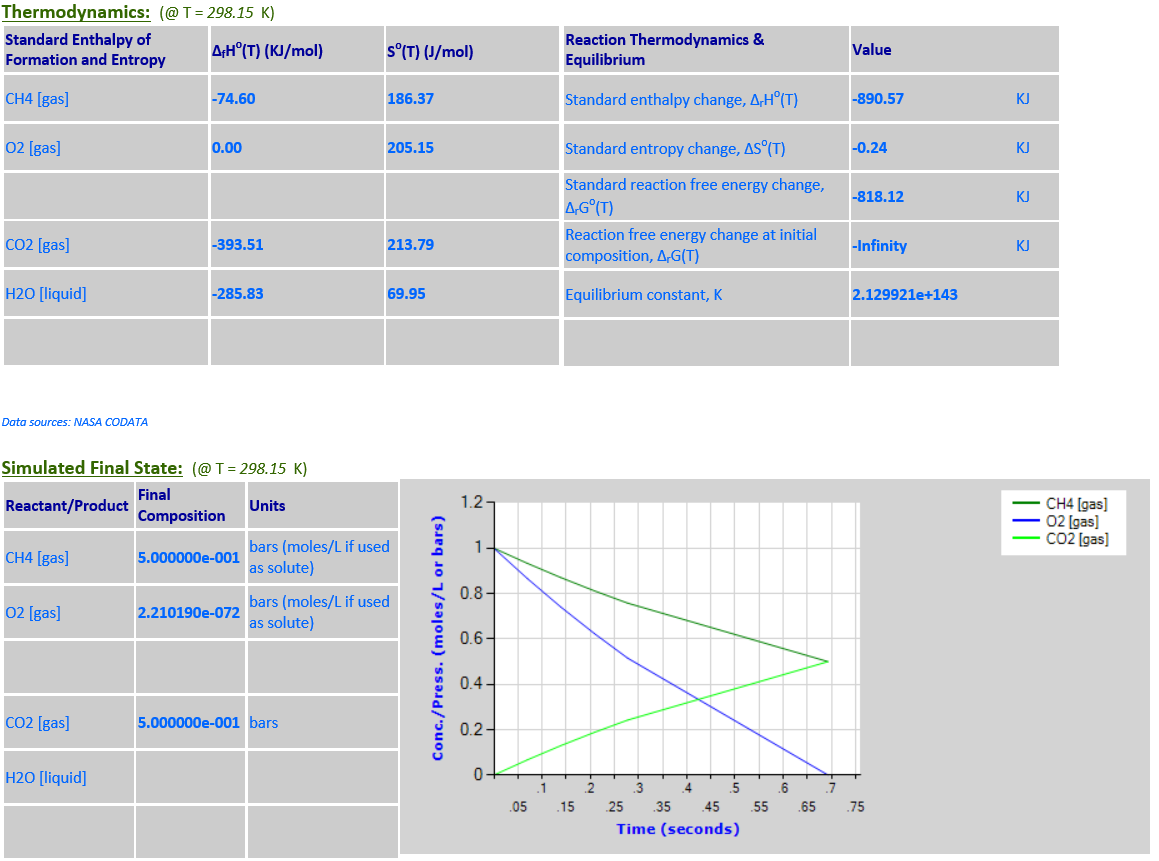
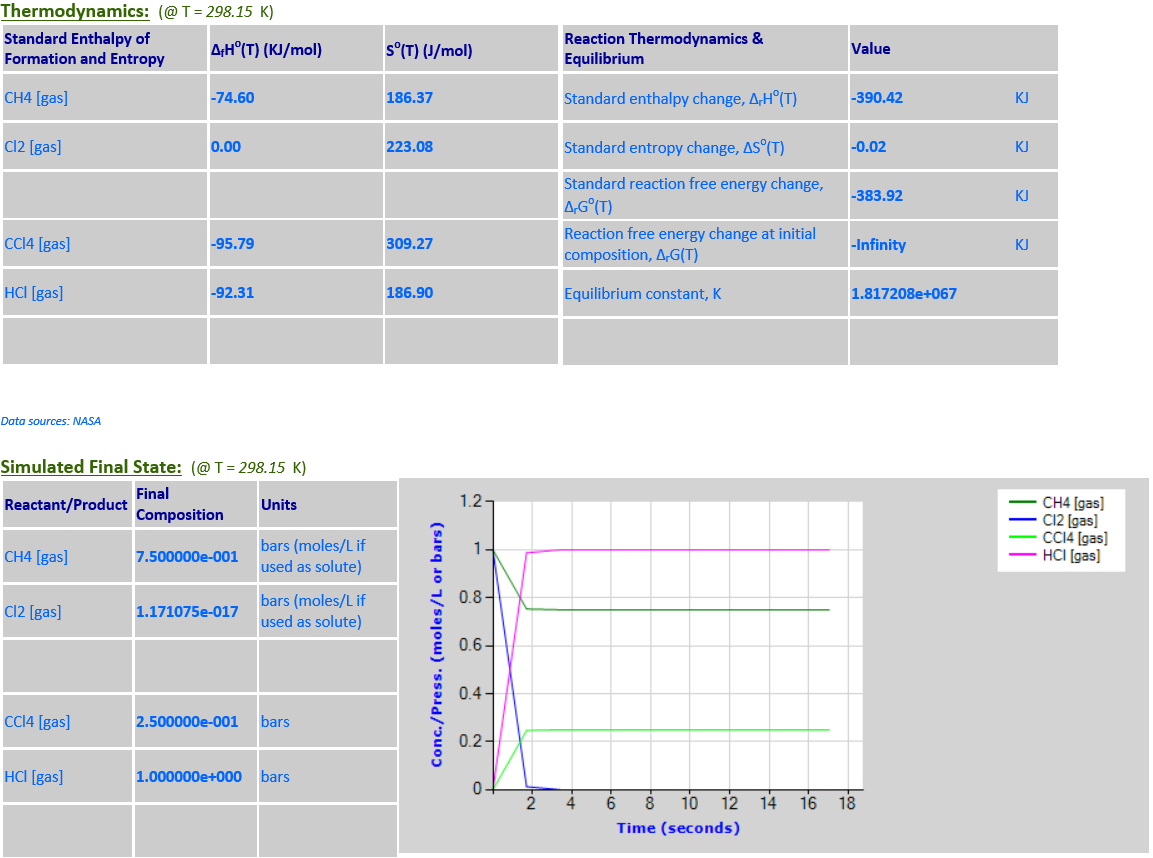
15. CH4 (g) +
4Cl2 (g) <--> CCl4 (g) + 4HCl (g)
·
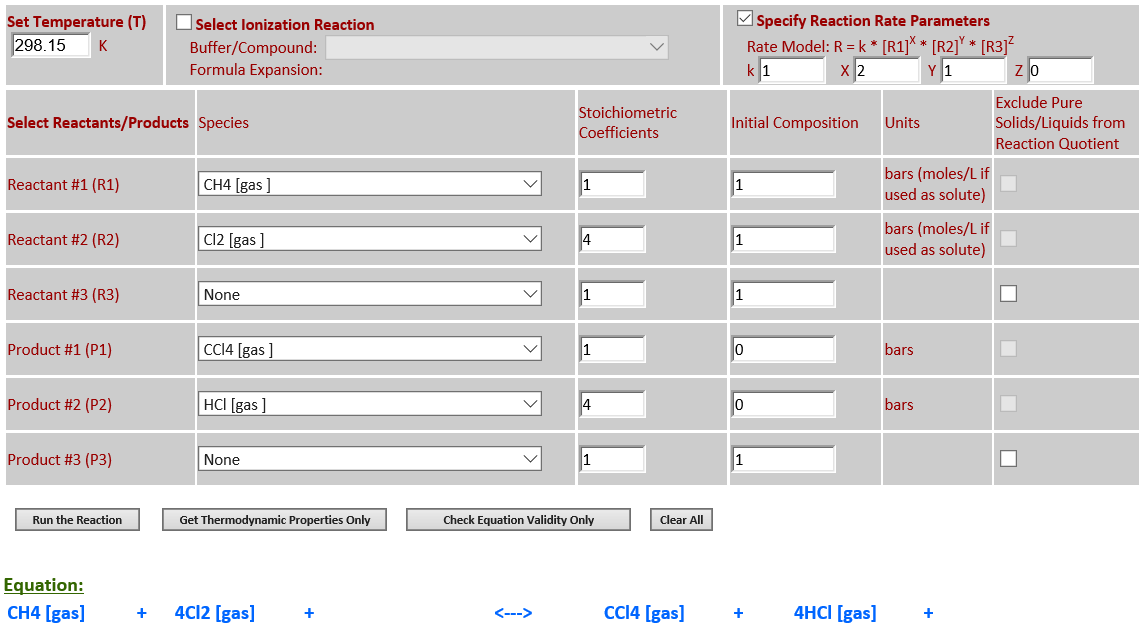
·
16. Ionization: HL (aq) <--> H+
(aq) + L- (aq) ;
HL=C2H4O2
·
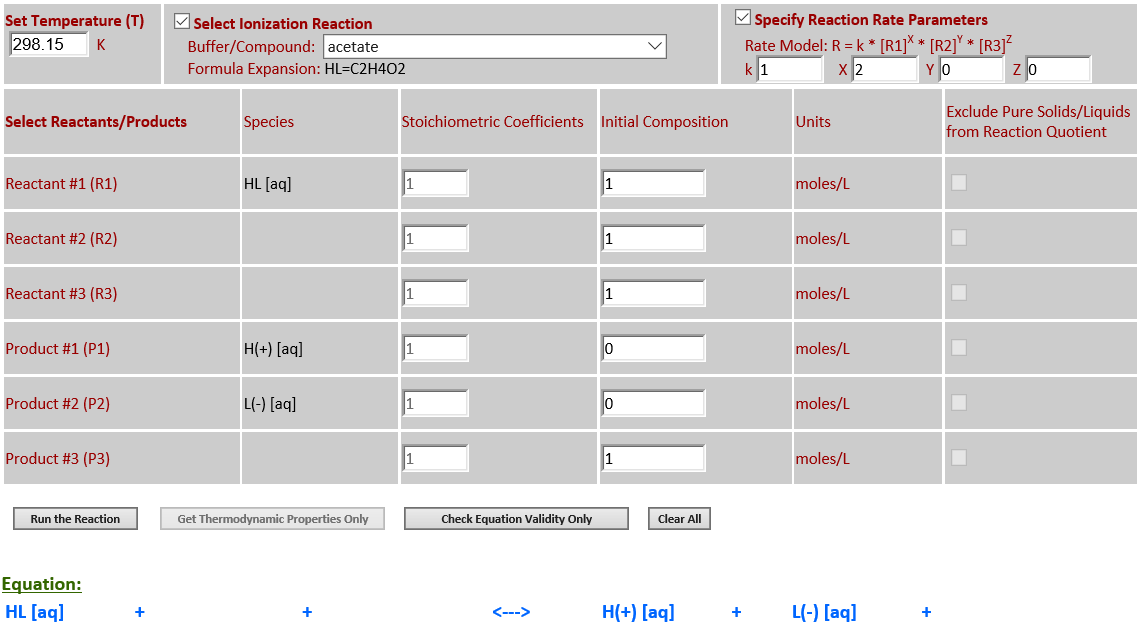
·
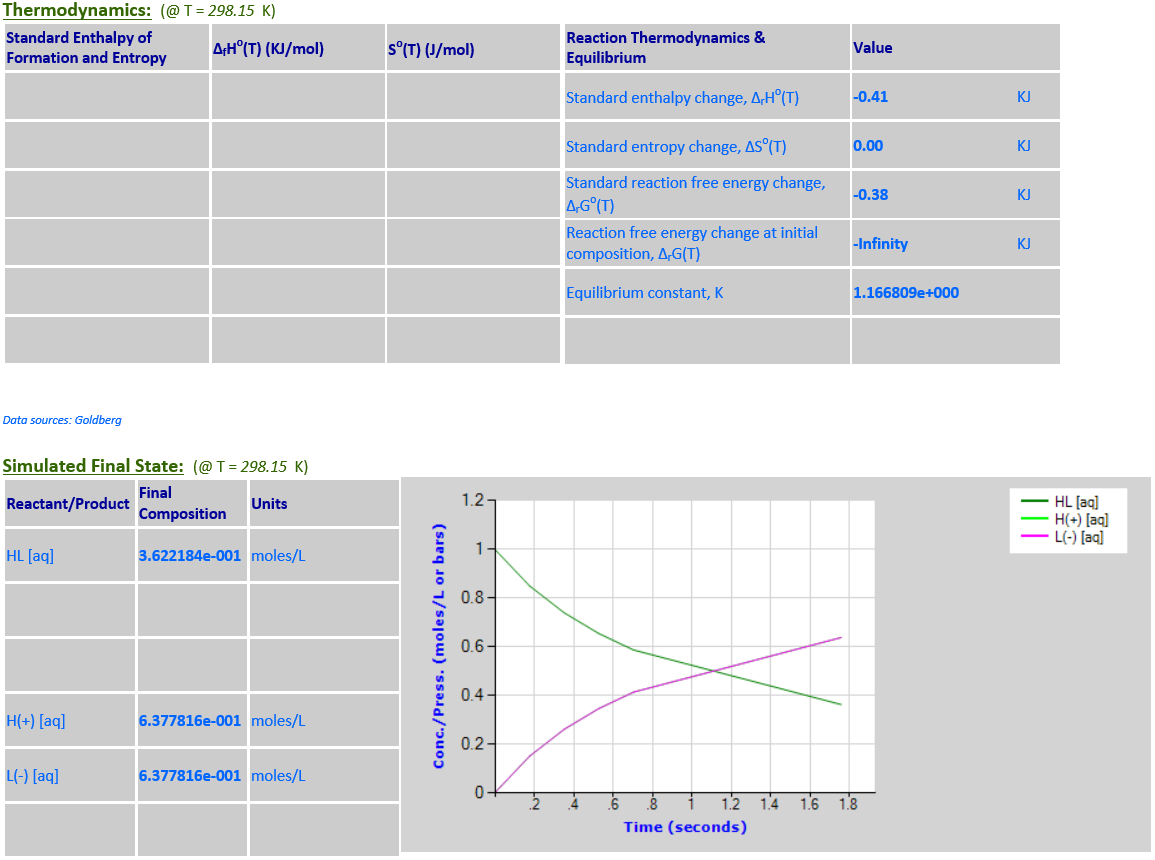
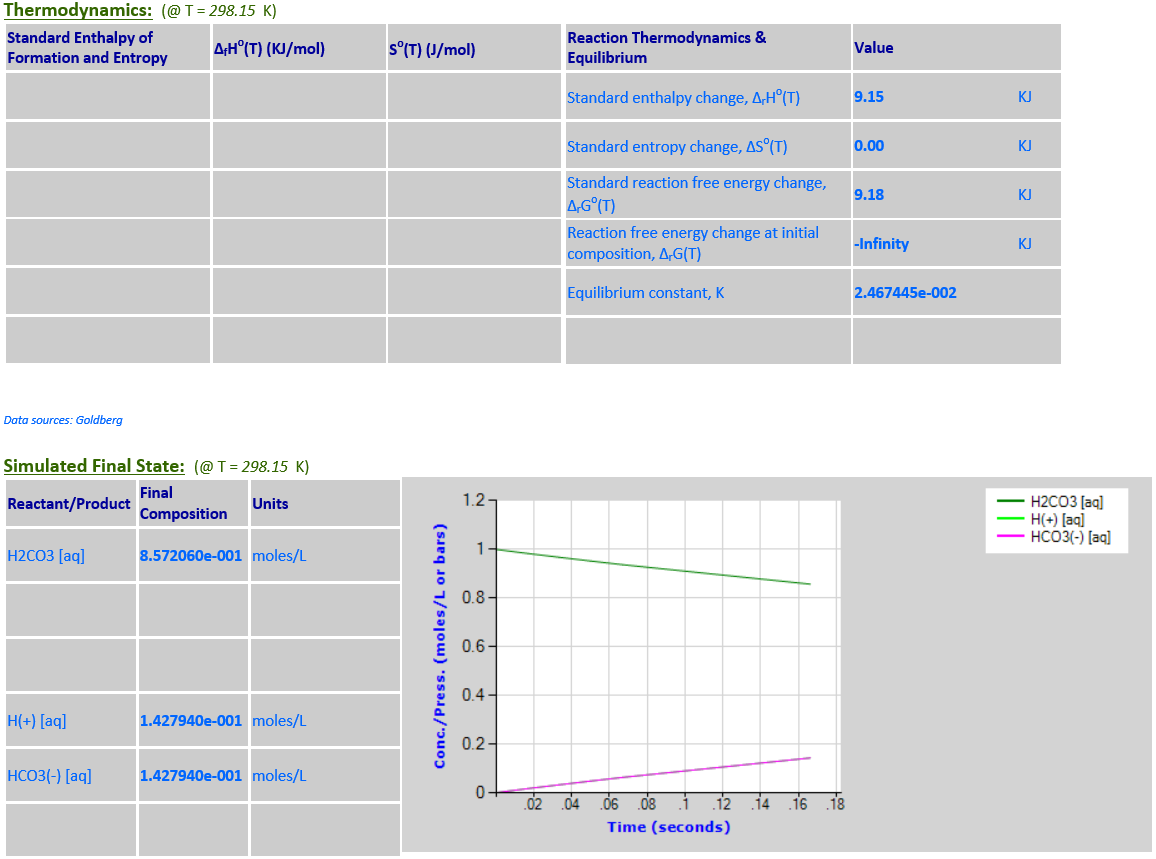
17. Ionization: H2CO3 (aq) <--> H+
(aq) + HCO3- (aq)
·
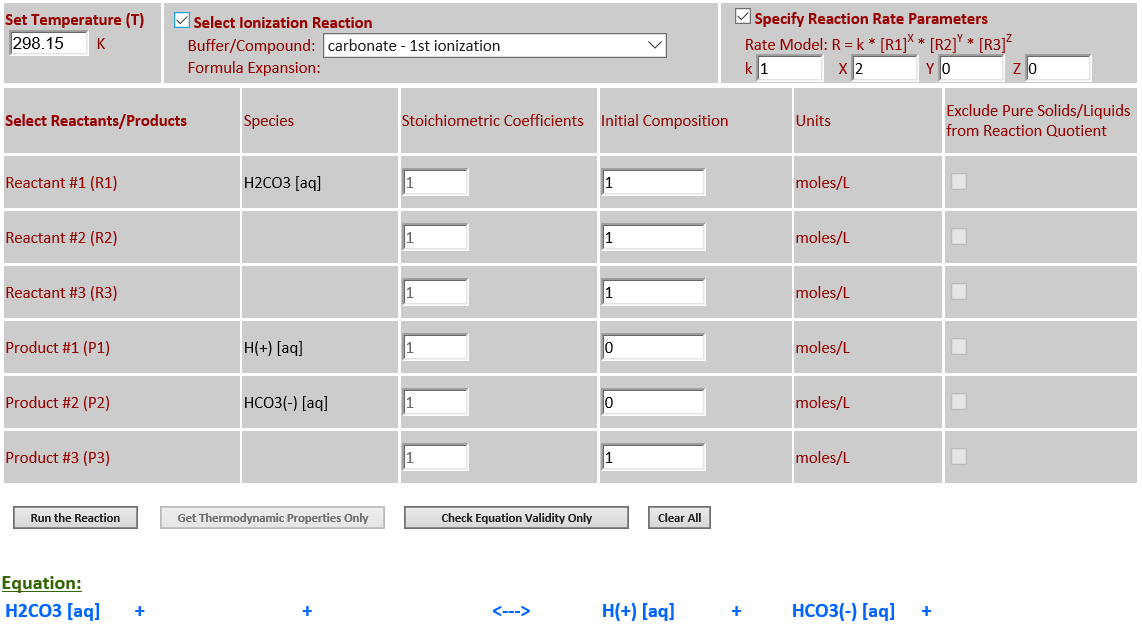
·
18. Reaction Rate Modeling:
H2 (g) + I2 (g) <--> 2HI (g) (zeroth order)
·
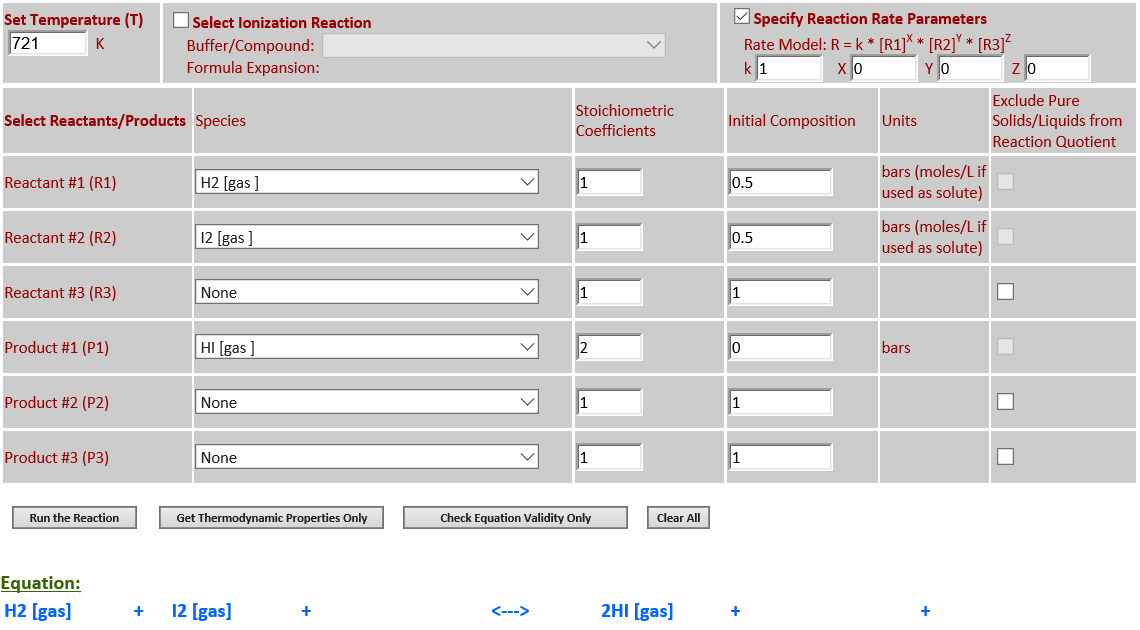
·
19. Reaction Rate Modeling:
H2 (g) + I2 (g) <--> 2HI (g) (first order)
·
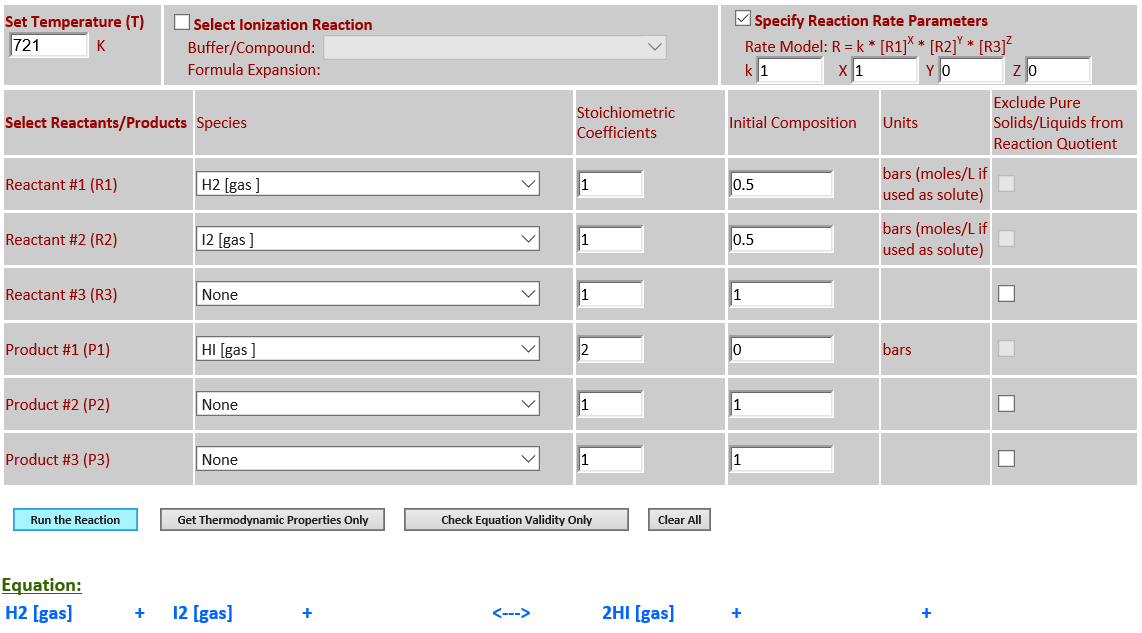
·
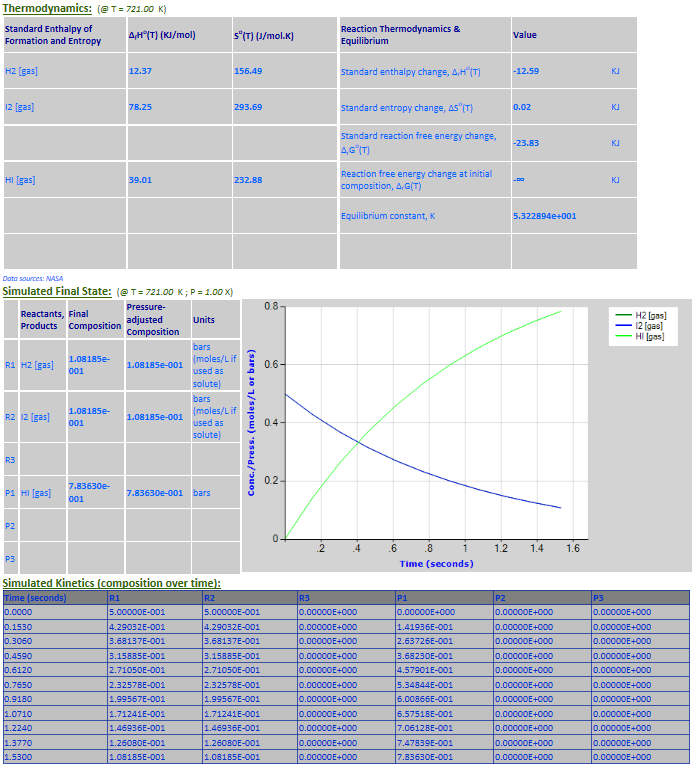
20. Reaction Rate Modeling:
H2 (g) + I2 (g) <--> 2HI (g) (second order)
·
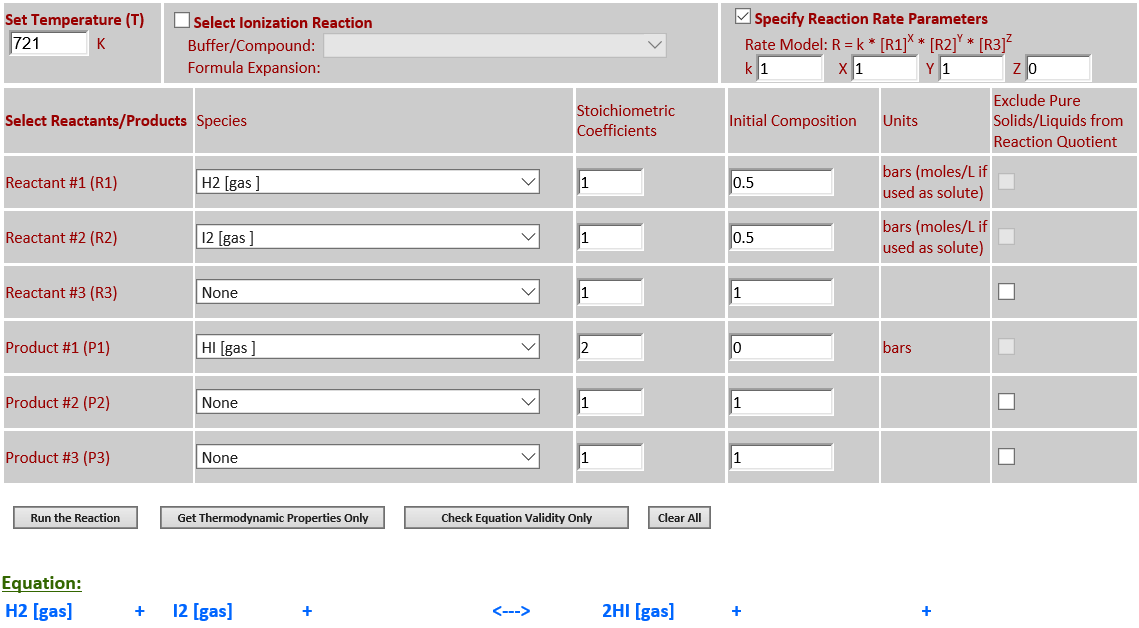
·
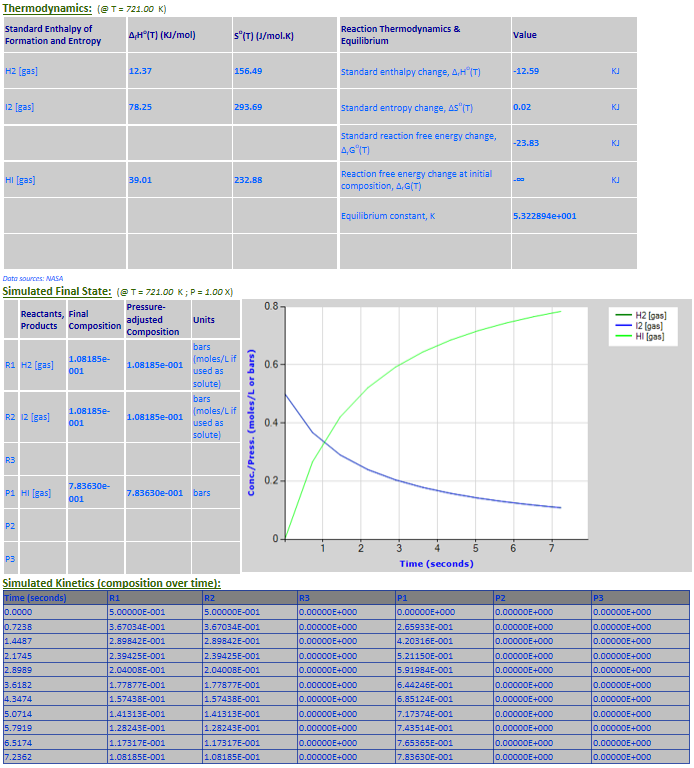
21. Reaction Rate Modeling:
H2 (g) + I2 (g) <--> 2HI (g) (second order)
·

·
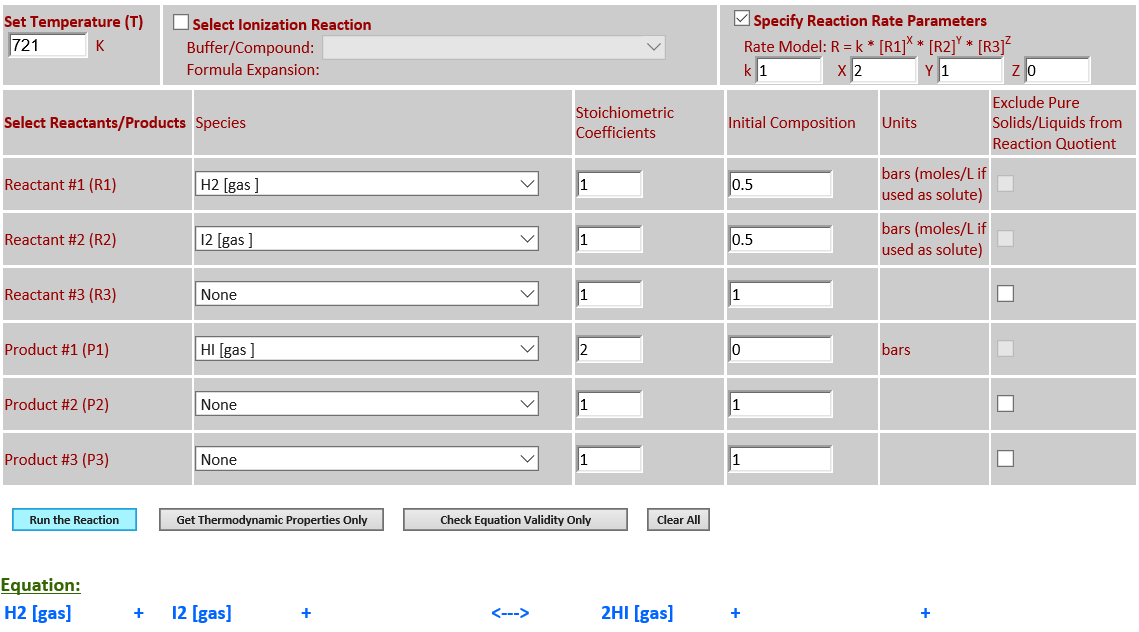
22.
Reaction Rate Modeling: H2 (g) +
I2 (g) <--> 2HI (g) (third order)
·
·
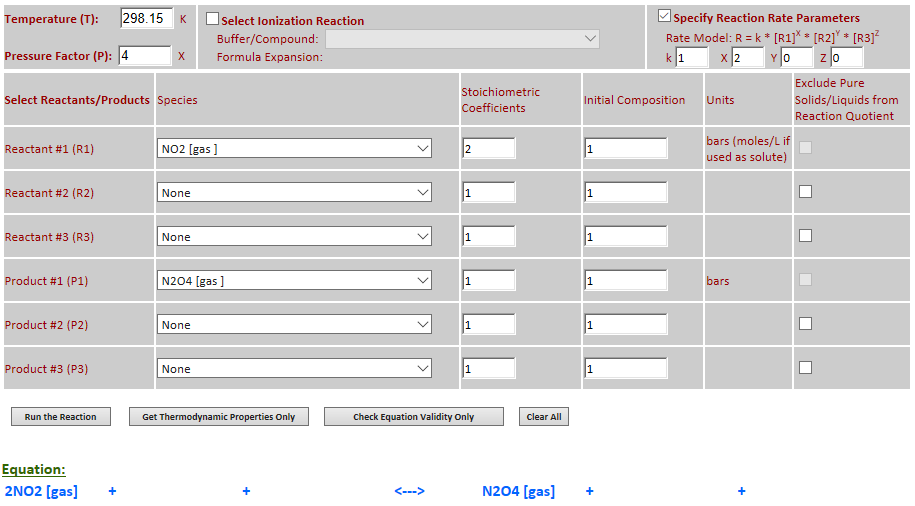
23.
Effect of pressure change on equilibrium: 2NO2 (g) <--> N2O4 (g)
·
Example 2 with pressure increased by a factor of 4. Equilibrium shifts to the
right.
·
·
·
Example 2 with pressure decreased by a factor of 4. Equilibrium shifts to the
left.
·
·
24.
Effect of pressure change on
equilibrium: H2 (g) + CO2 (g) <--> H2O (g) + CO (g)
·
Example 4 with pressure
increased by a factor of 4. Pressure changes have no effect on the equilibrium
since there are equal numbers of gas molecules on both sides of the reaction.
·
·
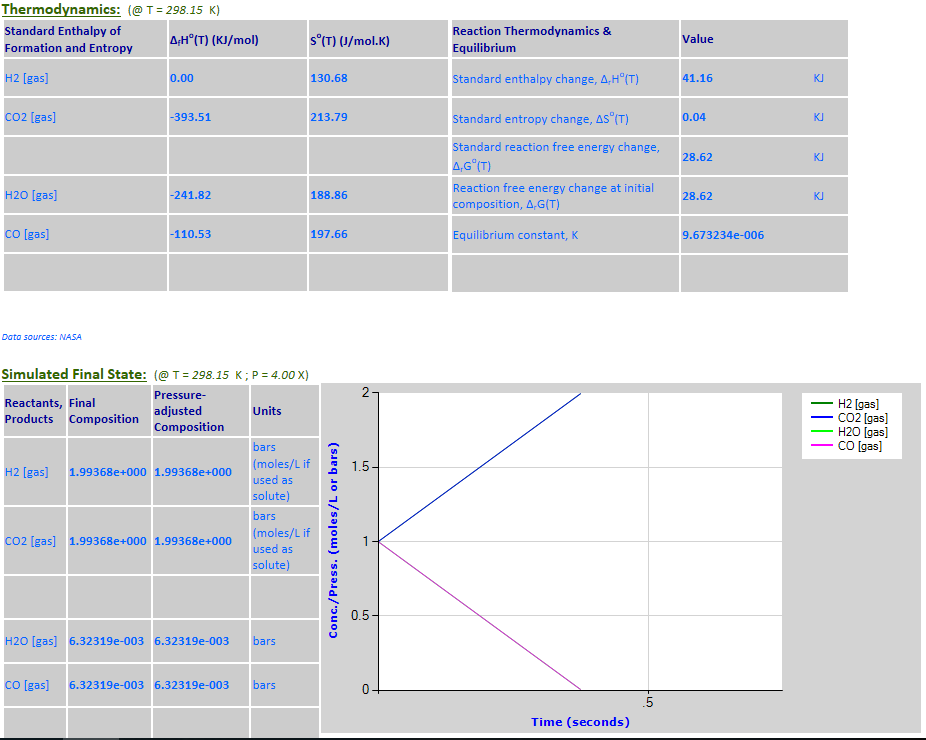
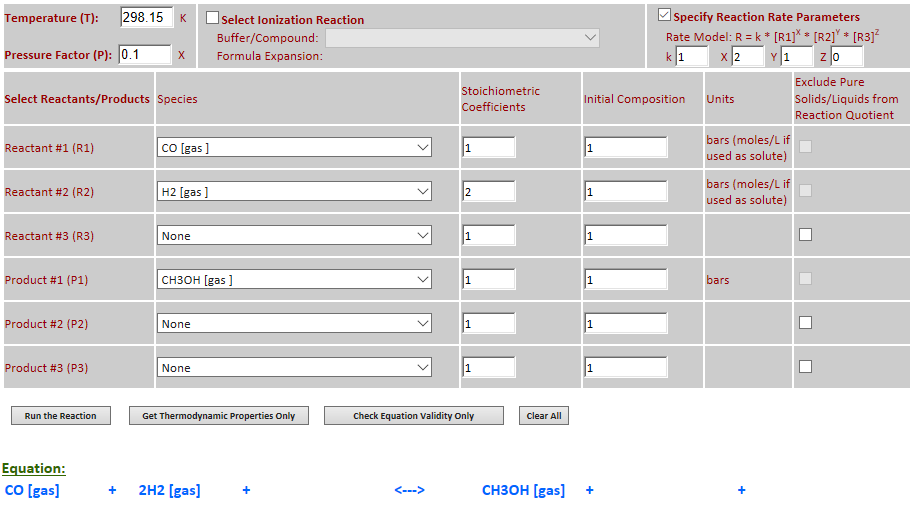
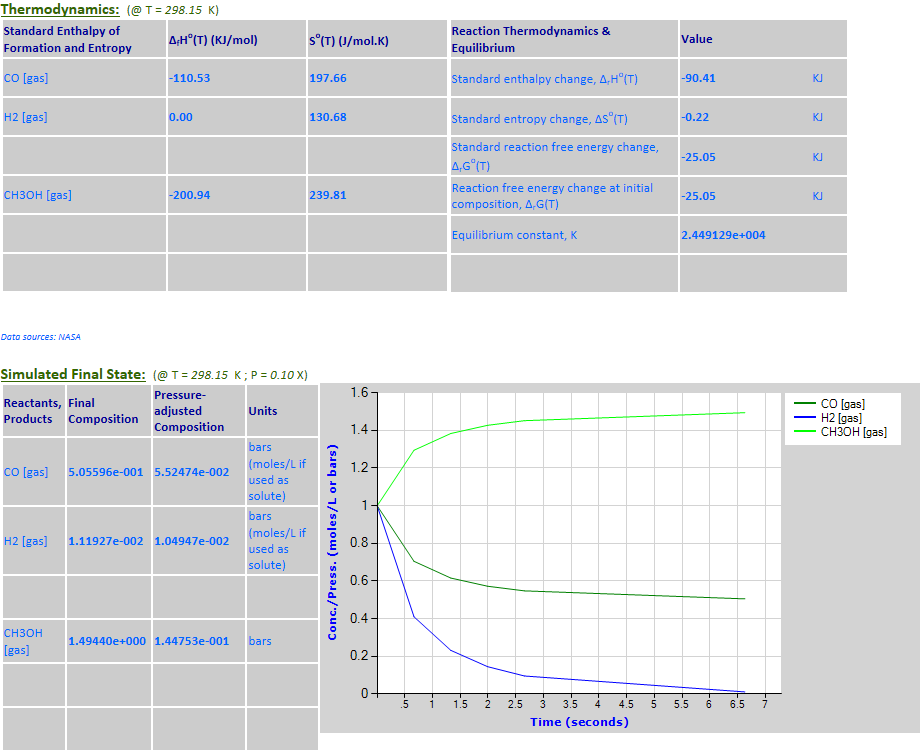
25.
Effect of pressure change on
equilibrium: CO (g) + 2H2 (g) <--> CH3OH (g)
·
Example 5 with pressure decreased by a factor of 10.
·
·
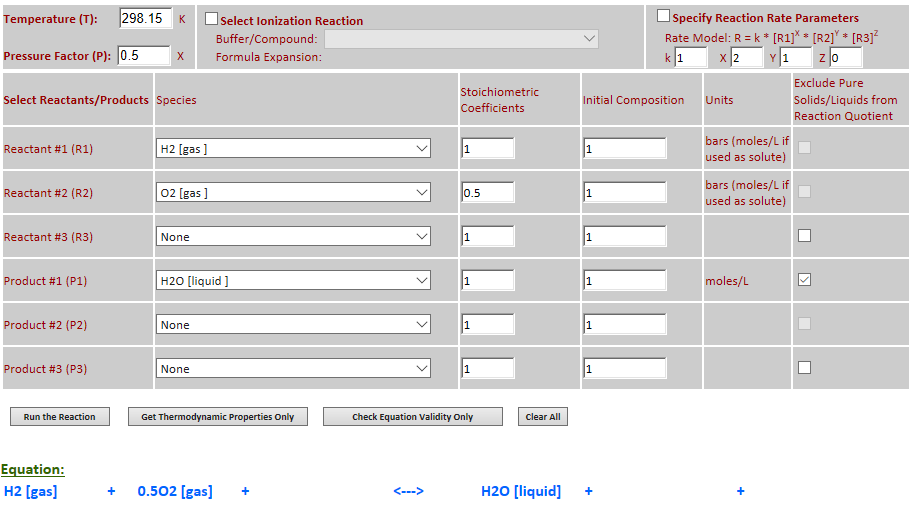
26.
Effect of pressure change on
equilibrium: H2 (g) + 0.5O2 (g) <--> H2O (l)
·
Example 6 with pressure decreased by a factor of 2.
·
·
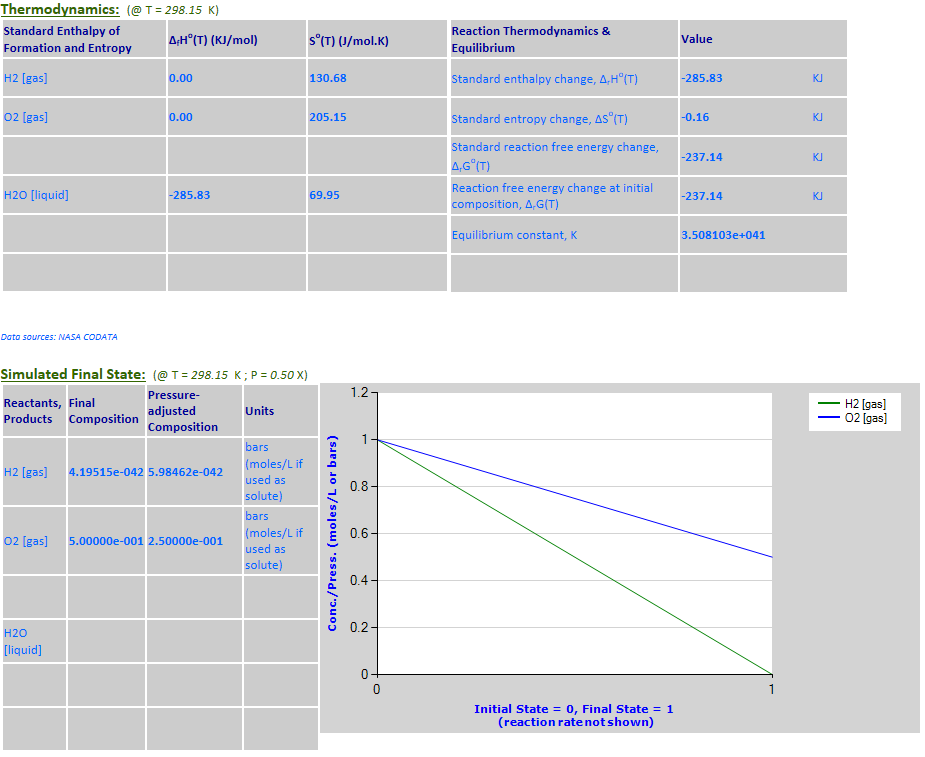
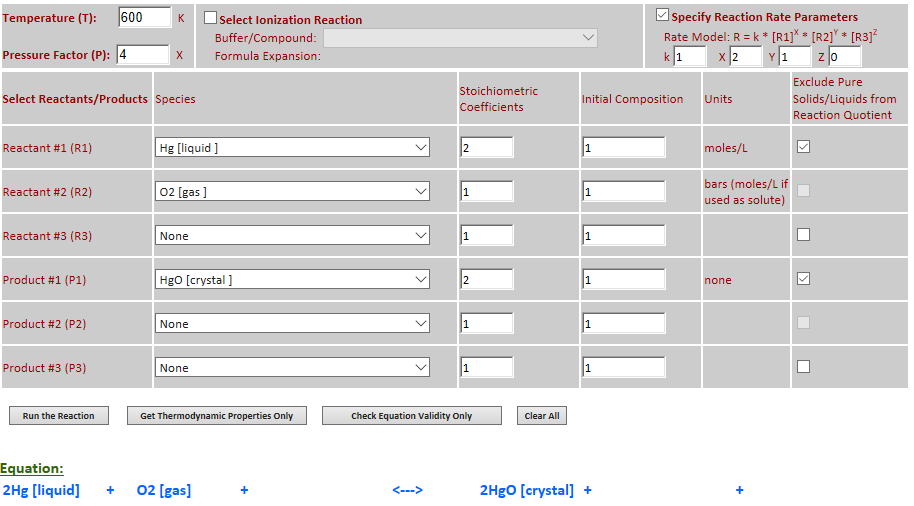
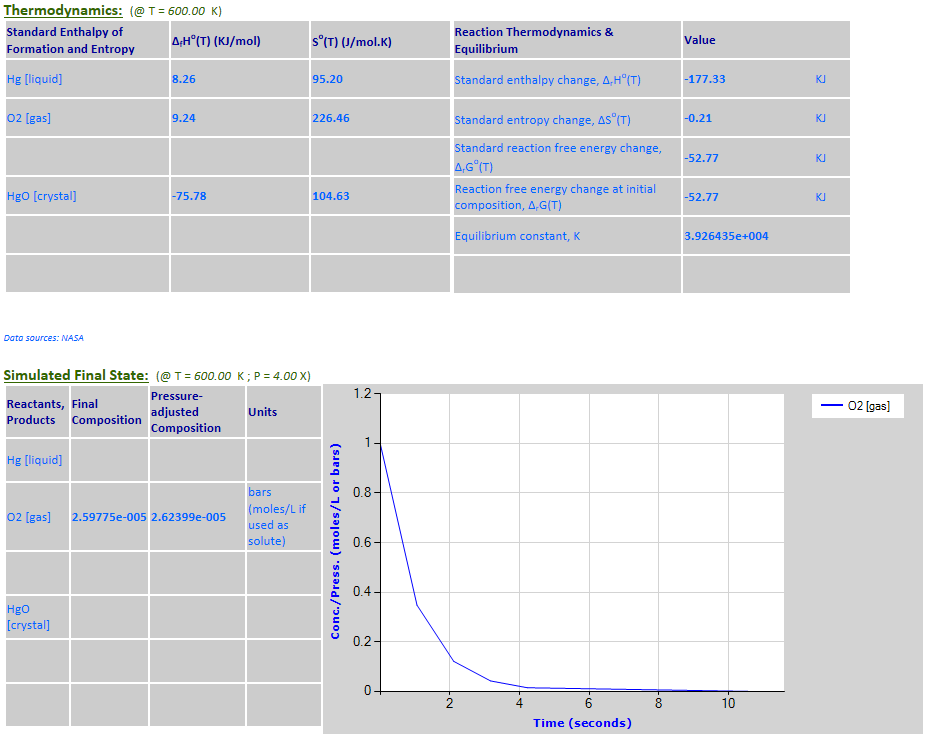
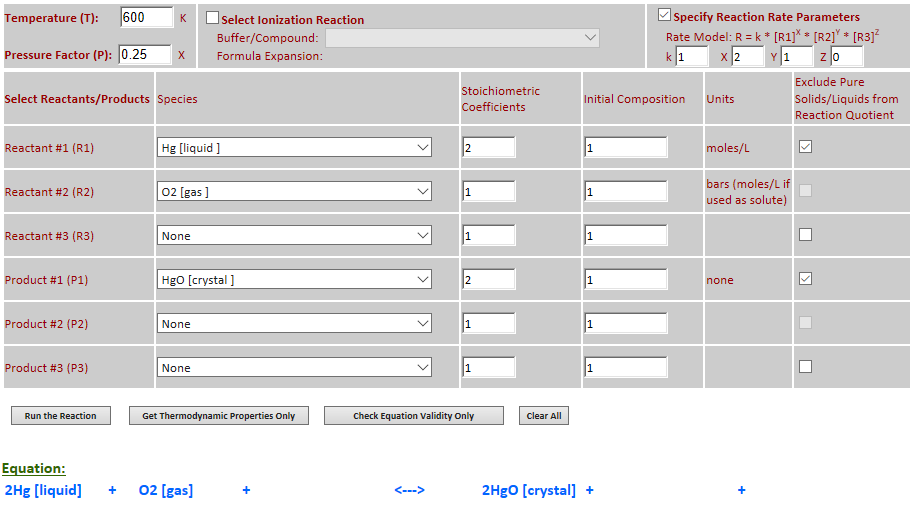
27.
Effect of pressure change on
equilibrium: 2Hg (l) + O2 (g) <--> 2HgO (c)
·
Example 7 with pressure increased by a factor of 4.
·
·
·
Example 7 with pressure decreased by a factor of 4.
·
·
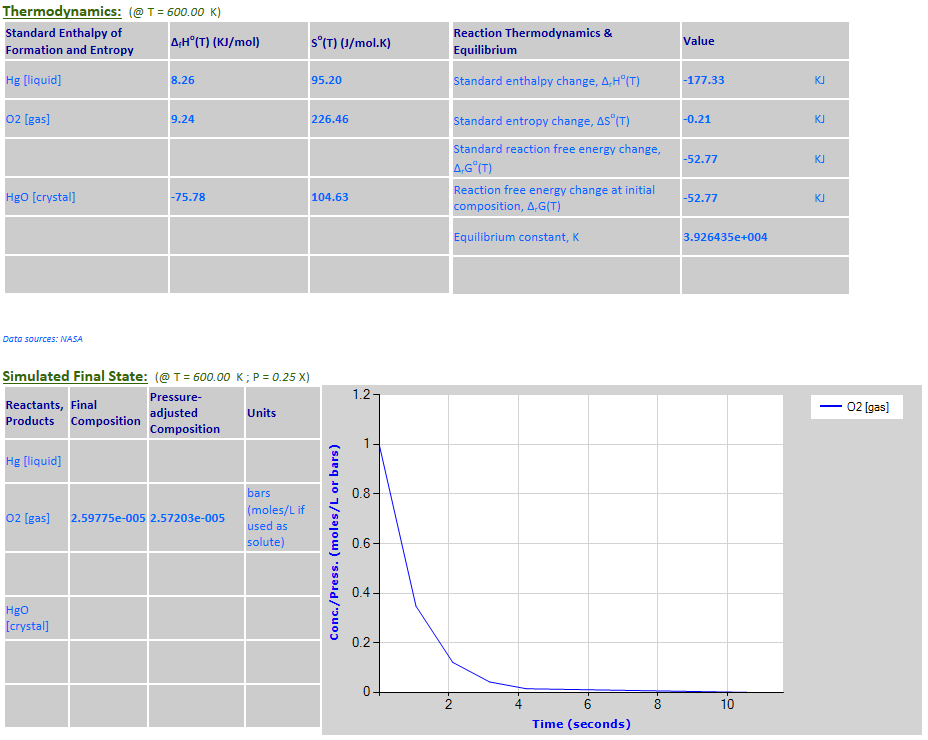
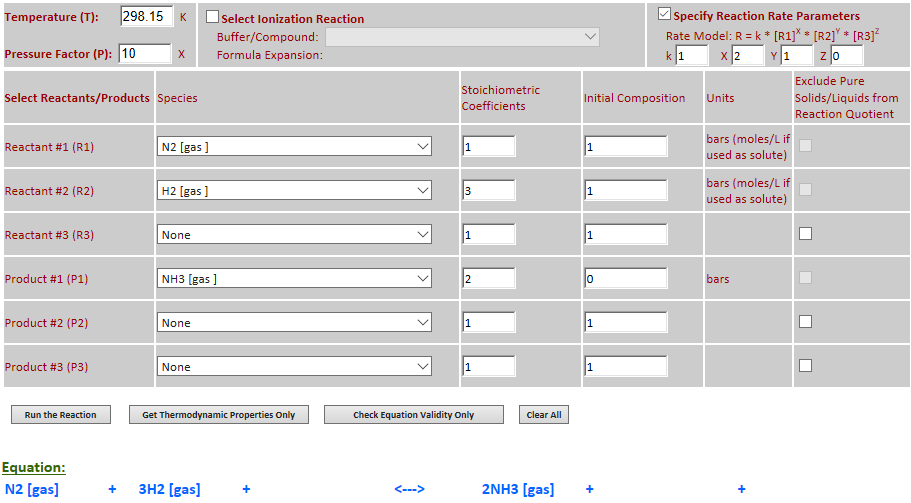
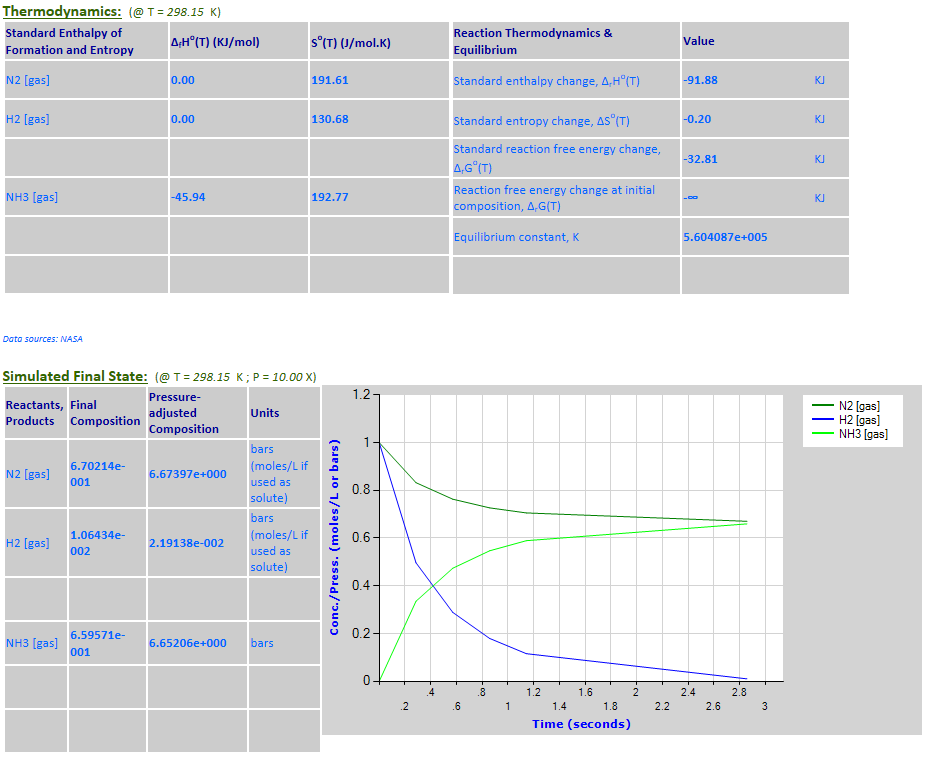
28.
Effect of pressure change on
equilibrium: N2 (g) + 3H2 (g) <--> 2NH3 (g)
·
Example 12 with pressure increased by a factor of 10.
·
·
·
Example 12 with pressure decreased by a factor of 10.
·
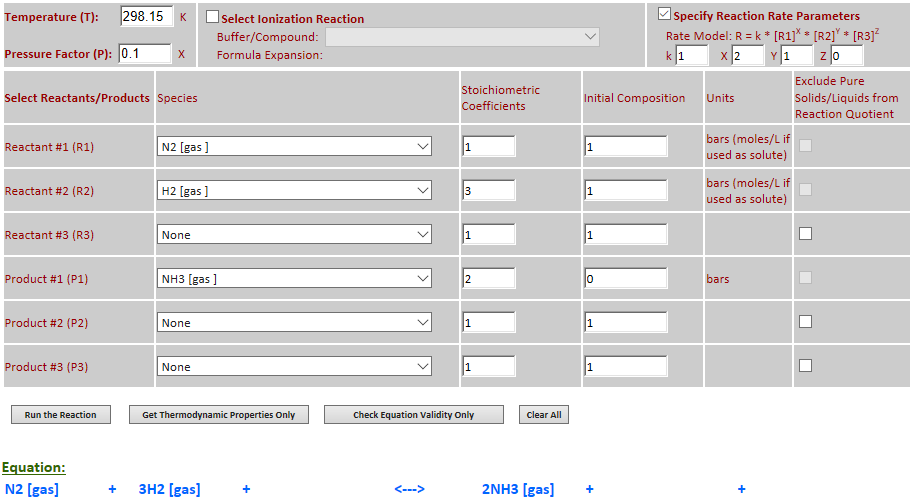
·